NM_000059.4:c.5661G>A (p.Thr1887=) is a synonymous variant in BRCA2 exon 11. BP1_Strong is met: the variant is a silent substitution outside both ENIGMA-defined clinically important functional domains (PALB2 binding aa 10-40; DNA binding aa 2481-3186), and SpliceAI predicts no splicing impact (max delta = 0.00).1 This variant is present in gnomAD v4.1 at ultra-low frequency (2/1,612,732 alleles, AF=1.24e-6) in the South Asian and European (non-Finnish) populations. It is absent from gnomAD v2.1. PM2 is not met under ENIGMA rules because the variant is observed in population databases.2 The variant has been classified as Likely Benign by the ENIGMA expert panel in ClinVar (Variation ID 51902). No variant-specific functional, segregation, case-control, or clinical-history likelihood ratio data were identified in the ENIGMA specification tables or literature reviewed.3 With only BP1_Strong (1 Strong Benign criterion) met and no other criteria triggered, the variant does not reach the ENIGMA Table 3 threshold for Likely Benign classification based solely on the evidence reviewed here. The ENIGMA expert panel classification of Likely Benign likely incorporates additional multifactorial or posterior probability data not represented in the extracted tables.4
BRCA2
Final classification
Likely Benign
BRCA2 c.5661G>A · p.Thr1887=
BRCA2
NM_000059.4:c.5661G>A (p.Thr1887=) is a synonymous variant in BRCA2 exon 11.
ENIGMA BRCA1/BRCA2 Specification v1.2 Table 3 combination rules applied to adjudicated criteria. BP1 met at Strong (Benign) strength: synonymous variant outside clinically important functional domains with no predicted splicing impact. BP6 met at Supporting (Benign) strength: ENIGMA expert panel classification in ClinVar. The combination of 1 Strong (Benign) + 1 Supporting (Benign) matches ENIGMA Table 3 Likely Benign rule. No pathogenic criteria are met, so no conflicting-evidence point system is required.
Classification rationale
BP1BP6
Likely Benign
BRCA2 c.5661G>A
BP1 + BP6
→
Likely Benign
Gene diagram
· NM_000059.4 · variants mapped to exon structure
BRCA2
NM_000059.4
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 12 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BP1
strong
Benign
c.5661G>A is a synonymous substitution (p.Thr1887=) located at amino acid position 1887, which is outside both ENIGMA-defined clinically important functional domains (PALB2 binding: aa 10-40; DNA binding: aa 2481-3186). SpliceAI predicts no splice impact (max delta = 0.00, ≤ 0.1). This satisfies all conditions for BP1_Strong under ENIGMA v1.2.
Synonymous variant p.Thr1887= outside functional domains. SpliceAI max delta = 0.00. Position 1887 is > 40 and < 2481confirming it is outside both clinically important functional domains.
Assessed · not applied
Pathogenic
PS3
c.5661G>A is not listed in ENIGMA Table 9 curated functional assay results.
PS4
No case-control data available.
PM2
Absent from gnomAD v2.1 (non-cancer, exome) but present in gnomAD v4.1 (2/1,612,732 alleles, AF=1.24e-6; 1 allele in South Asian, 1 in European non-Finnish).
PP1
No quantitative co-segregation data available for this variant.
PP3
ENIGMA PP3 requires either (a) missense/in-frame variant inside a clinically important functional domain with BayesDel no-AF ≥ 0.30, or (b) predicted splicing impact with SpliceAI ≥ 0.2.
PP4
Variant not found in PMID_31853058 BRCA2 clinical history LR table (2,182 rows searched by HGVS nucleotide).
Benign
BA1
ENIGMA BA1 requires FAF > 0.001 (0.1%) in gnomAD v2.1 or v3.1.
BS1
ENIGMA BS1_Strong requires FAF > 0.0001 (0.01%) in gnomAD v2.1 or v3.1.
BS2
ENIGMA BS2 requires observation in healthy adults without Fanconi Anemia phenotype, with point-based scoring.
BS3
c.5661G>A is not listed in ENIGMA Table 9 curated functional assay results for BRCA2 (4,734 rows searched).
BS4
No segregation or posterior probability data available.
BP5
Variant not found in PMID_31853058 BRCA2 clinical history LR table or HUMU-40-1557-s001 SuppT1.
N/A · 14
PVS1 · PS1 · PS2 · PM1 · PM3 · PM4 · PM5 · PM6 · PP2 · PP5 · BP2 · BP3 · BP4 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
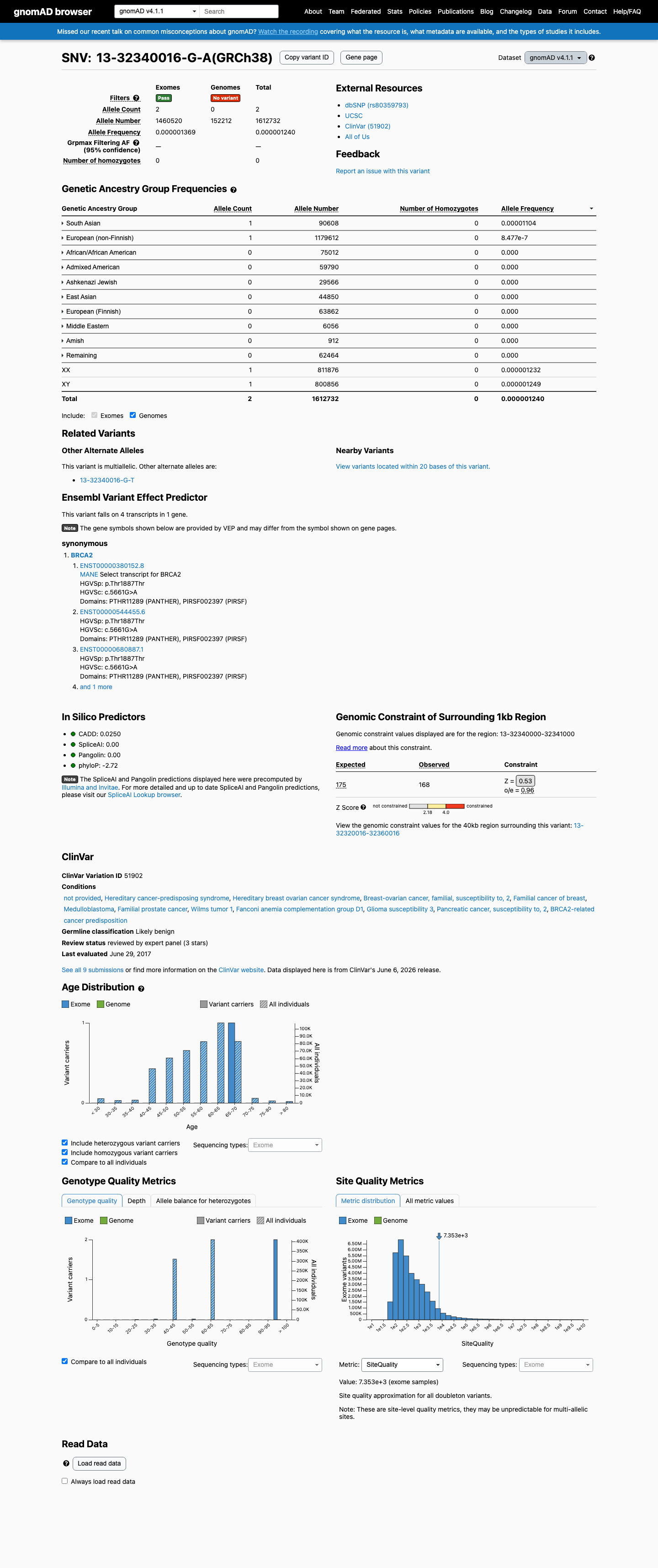
This variant is present in gnomAD v4.1 (AF= 1.24013e-06; MAF= 0.00012%, 2/1612732 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 1.10366e-05; MAF= 0.00110%, 1/90608 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00012%
· 2 / 1,612,732
0 hom
0 hom
South Asian 1 / 90,608 |
0.0011% |
European (non-Finnish) 1 / 1,179,612 |
8.5e-05% |
+ 8 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
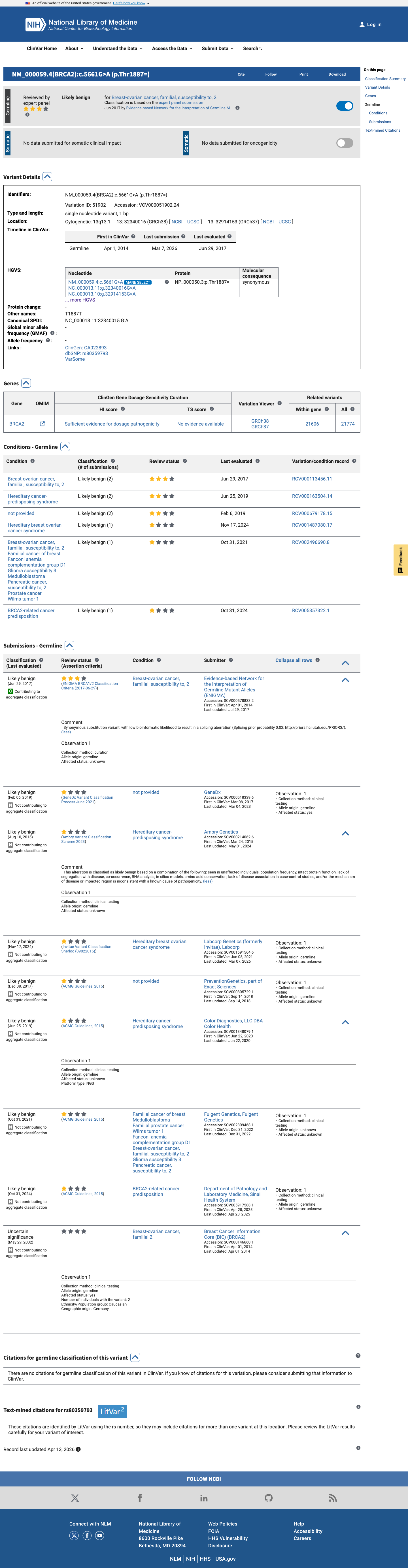
This variant has been reported in ClinVar as Likely benign (7 clinical laboratories) and as Uncertain significance (1 clinical laboratory) and as Likely benign by Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) (expert panel). (ClinVarID = 51902)
In silico
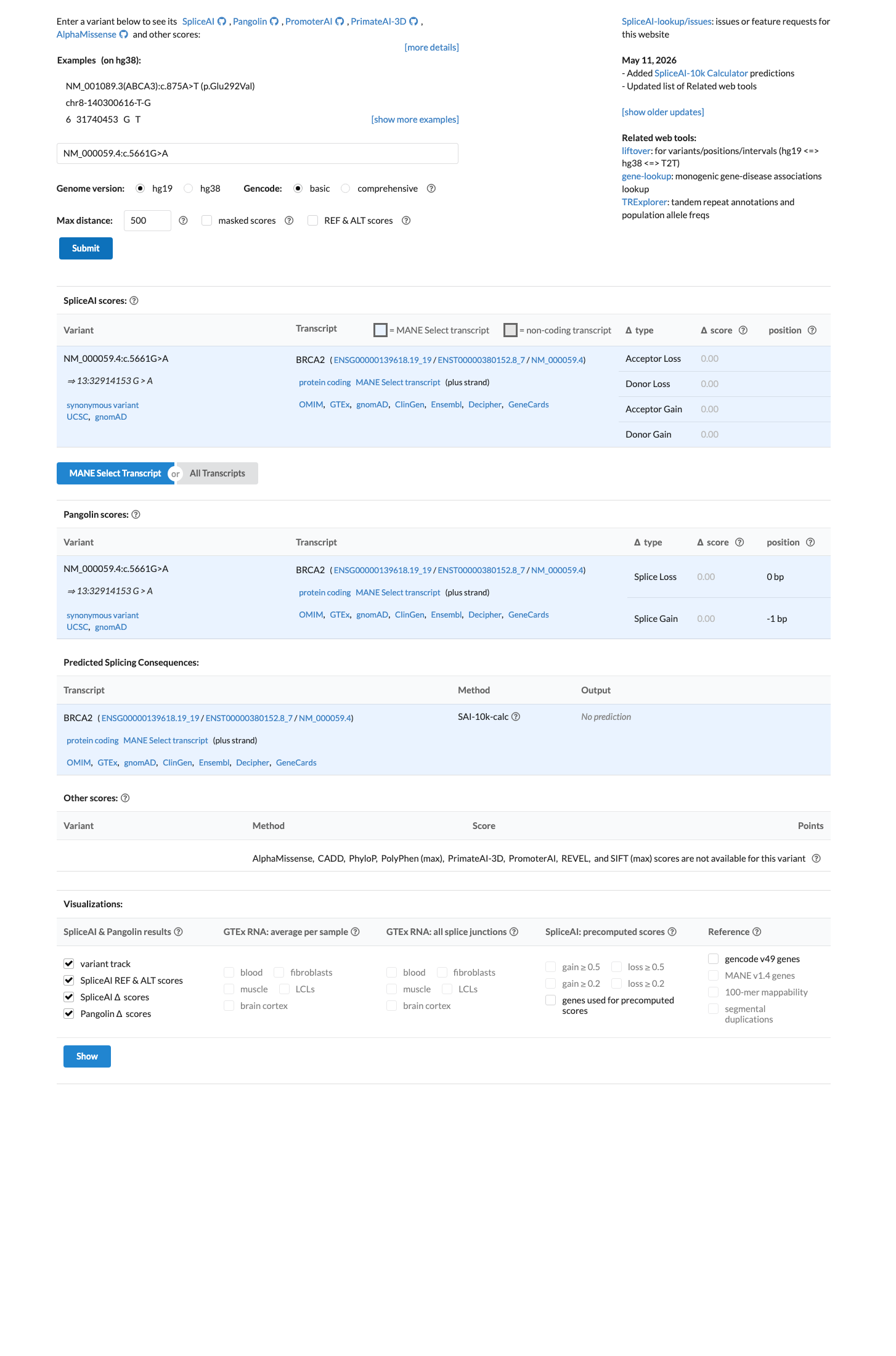
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 7 PMIDs not cited in assessment
17392385 ↗
American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography.
CLINVAR