BP1_Strong is met: c.750G>A is a silent (synonymous) substitution (p.Val250=) located outside both BRCA2 clinically important functional domains (PALB2 binding domain aa10-40 and DNA binding domain aa2481-3186), and SpliceAI predicts no splicing impact (max delta = 0.00, ≤ 0.1 threshold).1 BS1_Supporting is met: the gnomAD v2.1 non-cancer exome filter allele frequency (FAF) is 4.75e-05 (0.00475%), falling in the ENIGMA BS1_Supporting range (FAF > 0.002% and ≤ 0.01%). The highest population sub-frequency is in European (non-Finnish) at 7.77e-05 (10/128,766 alleles).2 PM2_Supporting is not met because the variant is present in gnomAD population databases (11 alleles in v2.1, 134 alleles in v4.1), failing the ENIGMA requirement of absence from outbred population controls.3 PP3 is not met: SpliceAI max delta is 0.00 (< 0.2) and the variant is outside clinically important functional domains, failing both PP3 application pathways in the ENIGMA specification.4 BA1 (Stand-Alone Benign) is not met: gnomAD grpmax FAF of 4.75e-05 (v2.1) and 9.28e-05 (v4.1) are both well below the 0.1% (FAF > 0.001) threshold.5 BS3, BS4, PP1, PP4, BP5, and BP7 are not assessed due to absence of variant-specific functional, segregation, or clinical-history LR data in the ENIGMA VCEP-curated datasets (Table 9, clinical_history_LR.xlsx, HUMU-40-1557-s001, ST7).6 PVS1, PS1, PS2, PS3, PS4, PS5, PM1, PM5, PM6, PP2, PP5, BP2, BP3, BP6 are not applicable for this synonymous variant under the ENIGMA VCEP framework.7 Under the ENIGMA Table 3 combination rules, 1 Strong Benign criterion (BP1_Strong) plus 1 Supporting Benign criterion (BS1_Supporting) reaches the threshold for Likely Benign. Under the ENIGMA point system: BP1_Strong = -4 points, BS1_Supporting = -1 point, total = -5 points (Likely Benign range: -6 to -2).8 ClinVar lists this variant as Likely Benign with expert panel review status by ENIGMA (ClinVar ID 135814), with 10 clinical laboratories reporting Likely Benign and 5 reporting Benign. This clinical consensus is concordant with the ENIGMA framework-based assessment.9
BRCA2
Final classification
Likely Benign
BRCA2 c.750G>A · p.Val250=
BRCA2
BP1_Strong is met: c.750G>A is a silent (synonymous) substitution (p.Val250=) located outside both BRCA2 clinically important functional domains (PALB2 binding domain aa10-40 and DNA binding domain aa2481-3186), and SpliceAI predicts no splicing impact (max delta = 0.00, ≤ 0.1 threshold).
ENIGMA BRCA1/BRCA2 VCEP v1.2.0 Table 3 combination rules applied to adjudicated criteria. One Strong Benign criterion (BP1_Strong) plus two Supporting Benign criteria (BS1_Supporting, BP6_Supporting) independently satisfy multiple Likely Benign rules: 1 Strong (Benign) + 1 Supporting (Benign) and 2 Supporting (Benign). No pathogenic criteria are met; no conflicting evidence present. ENIGMA point system (conflicting_evidence only) confirms: BP1_Strong (-4) + BS1_Supporting (-1) + BP6_Supporting (-1) = -6, within Likely Benign range (-6 to -2). ClinVar expert panel (ENIGMA) also reports Likely Benign (ClinVar ID 135814).
Classification rationale
BS1BP1BP6
Likely Benign
BRCA2 c.750G>A
BS1 + BP1 + BP6
→
Likely Benign
Gene diagram
· NM_000059.4 · variants mapped to exon structure
BRCA2
NM_000059.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 13 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
supporting
Benign
BS1_Supporting per ENIGMA is met. The filter allele frequency (FAF) in gnomAD v2.1 (non-cancer, exome only) is 4.75e-05 (0.00475%), which falls in the range FAF > 0.00002 (0.002%) and ≤ 0.0001 (0.01%). The highest population sub-frequency is in European (non-Finnish) at AF=7.77e-05 (10/128,766 alleles, 0 homozygotes). The variant is also present in gnomAD v4.1 with grpmax FAF 9.28e-05, consistent with the BS1_Supporting range. This frequency is higher than expected for a highly penetrant pathogenic BRCA2 variant.
gnomAD v2.1 exome non-cancer grpmax FAF = 4.75e-05 (0.00475%)within BS1_Supporting range (>0.002% and ≤0.01%)10/128
✓
BP1
strong
Benign
BP1_Strong per ENIGMA is met. c.750G>A is a silent (synonymous) substitution located at Val250, which is outside both BRCA2 clinically important functional domains (PALB2 binding domain aa10-40 and DNA binding domain aa2481-3186). SpliceAI predicts no splicing impact (max delta score = 0.00, well below the 0.1 threshold). The ENIGMA specification states: 'Apply BP1_Strong for silent substitution, missense or in-frame insertion, deletion or delins variants outside a (potentially) clinically important functional domain AND no splicing predicted (SpliceAI ≤ 0.1).' All conditions are satisfied.
Silent (synonymous) substitution p.Val250=V250 is outside BRCA2 clinically important domains (PALB2: aa10-40DNA binding: aa2481-3186)
Assessed · not applied
Pathogenic
PS3
No well-established functional studies demonstrating a damaging effect were identified.
PS4
No case-control studies demonstrating enrichment of c.750G>A in affected individuals were identified.
PM2
PM2_Supporting per ENIGMA requires the variant to be absent from gnomAD v2.1 (non-cancer, exome only) AND gnomAD v3.1 (non-cancer).
PP1
No co-segregation data were identified for c.750G>A.
PP3
PP3 per ENIGMA requires either (a) missense/in-frame variant inside a clinically important functional domain with BayesDel no-AF ≥ 0.30, or (b) SpliceAI ≥ 0.2 for any variant type regardless of location.
PP4
PP4 per ENIGMA requires a multifactorial likelihood ratio (LR ≥ 2.08) from calibrated clinical data.
Benign
BA1
BA1 (Stand-Alone Benign) per ENIGMA requires filter allele frequency (FAF) > 0.001 (0.1%) in gnomAD non-cancer, non-founder populations.
BS2
BS2 per ENIGMA requires observation in the absence of Fanconi Anemia (FA) phenotype features, with a point system per proband (Specifications Table 8).
BS3
BS3 per ENIGMA requires well-established functional studies showing no damaging effect, with code recommendations from calibrated published assays in Specifications Table 9.
BS4
BS4 per ENIGMA requires quantitative co-segregation analysis showing lack of segregation (LR ≤ 0.48).
BP4
BP4 per ENIGMA for silent variants applies only when the variant is inside a clinically important functional domain AND no splicing impact is predicted (SpliceAI ≤ 0.1).
BP5
BP5 per ENIGMA requires a multifactorial likelihood ratio (LR ≤ 0.48) from calibrated clinical data.
BP7
BP7 per ENIGMA can be applied at Strong strength for silent variants with well-established mRNA assay data showing no damaging effect, or at Supporting strength for silent variants inside a clinically important domain if BP4 is met.
N/A · 10
PVS1 · PS1 · PS2 · PM1 · PM5 · PM6 · PP2 · PP5 · BP2 · BP3
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
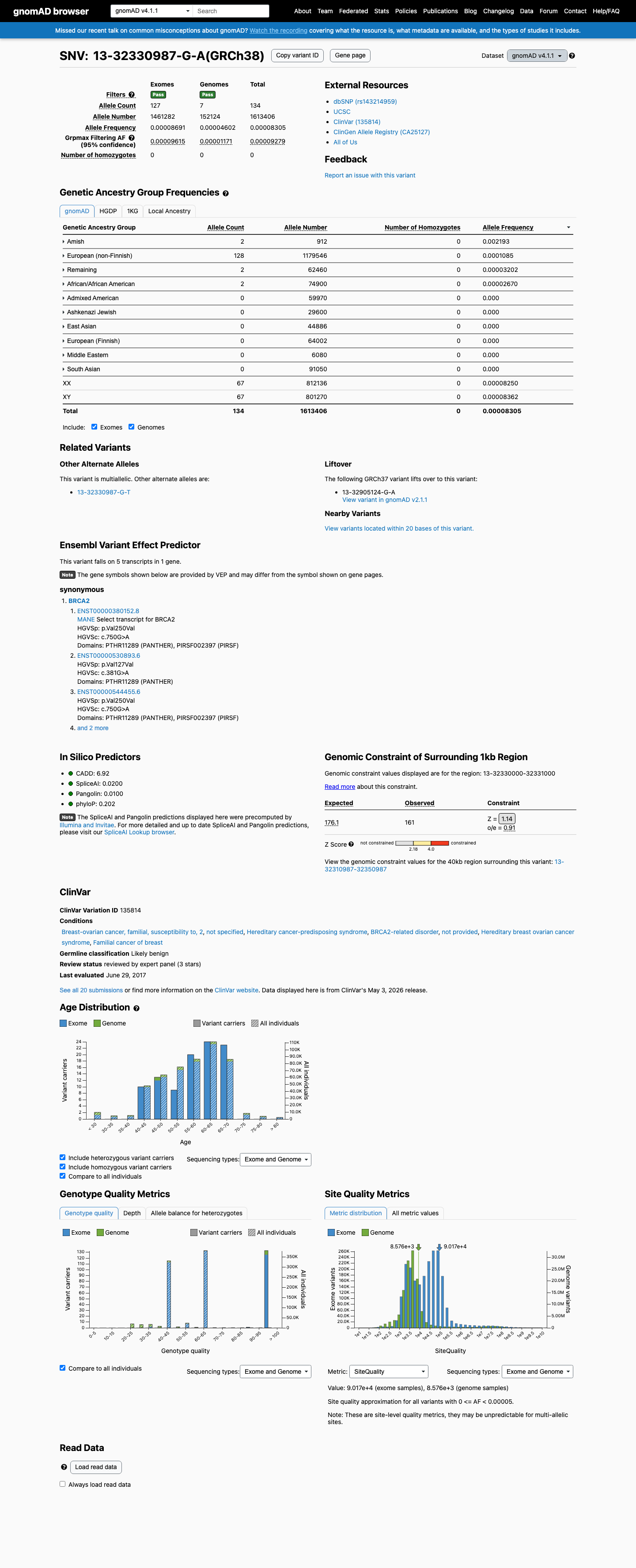
This variant is present in gnomAD v4.1 (AF= 8.30541e-05; MAF= 0.00831%, 134/1613406 alleles, homozygotes = 0) and has highest observed frequency in the Amish population (AF= 0.00219298; MAF= 0.21930%, 2/912 alleles, homozygotes = 0); grpmax FAF= 9.279e-05.
v2.1
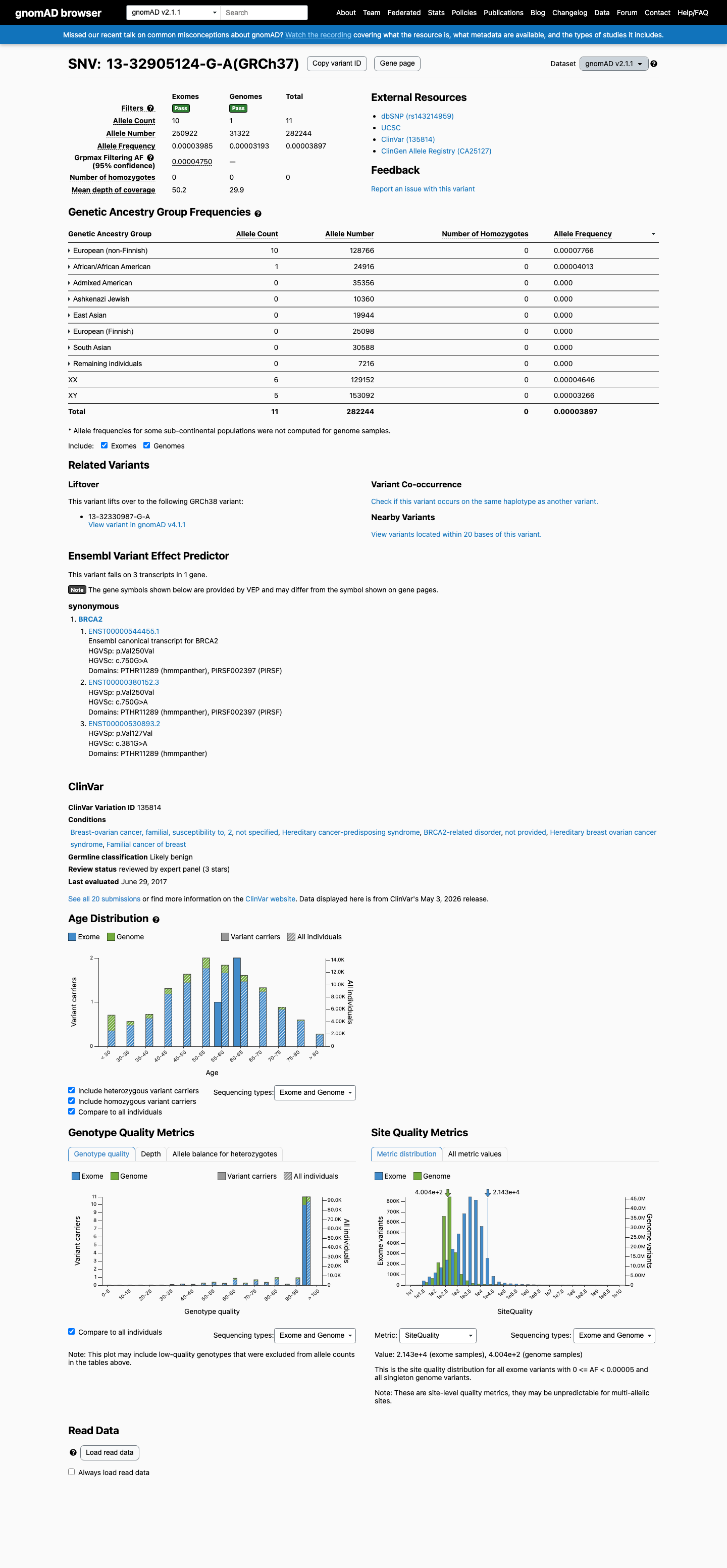
This variant is present in gnomAD v2.1 (AF= 3.89734e-05; MAF= 0.00390%, 11/282244 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 7.76603e-05; MAF= 0.00777%, 10/128766 alleles, homozygotes = 0); grpmax FAF= 4.75e-05.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0083%
· 134 / 1,613,406
0 hom · FAF 0.0093%
0 hom · FAF 0.0093%
Amish 2 / 912 |
0.22% |
European (non-Finnish) 128 / 1,179,546 |
0.011% |
Remaining individuals 2 / 62,460 |
0.0032% |
African/African American 2 / 74,900 |
0.0027% |
+ 6 not observed (Admixed American, European (Finnish), East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0039%
· 11 / 282,244
0 hom · FAF 0.0047%
0 hom · FAF 0.0047%
European (non-Finnish) 10 / 128,766 |
0.0078% |
African/African American 1 / 24,916 |
0.004% |
+ 6 not observed (Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
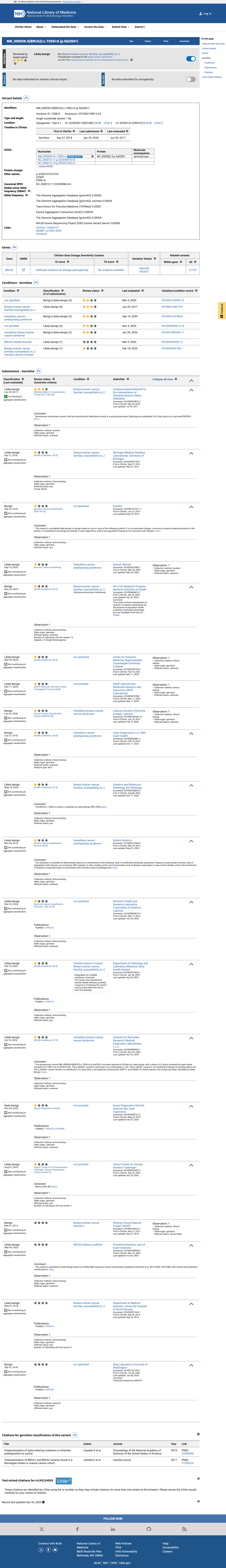
This variant has been reported in ClinVar as Likely benign (10 clinical laboratories) and as Benign (5 clinical laboratories) and as Likely Benign (1 clinical laboratory) and as likely benign (1 clinical laboratory) and as Likely benign by Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) (expert panel). (ClinVarID = 135814)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
27495310 ↗
Characterization of BRCA1 and BRCA2 variants found in a Norwegian breast or ovarian cancer cohort.
CLINVAR
31843900 ↗
Characterization of splice-altering mutations in inherited predisposition to cancer.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR