NM_000169.3:c.937G>T (p.Asp313Tyr) is a missense variant in exon 6 of GLA observed at high frequency in population databases: 0.304% in gnomAD v2.1 (624/205,260 alleles, 3 homozygotes) and 0.378% in gnomAD v4.1 (4,565/1,207,947 alleles, 10 homozygotes).1 Well-established functional studies demonstrate D313Y is a pseudodeficiency allele retaining approximately 60% of wild-type alpha-galactosidase A activity with proper lysosomal localization. The enzyme is stable at lysosomal pH, and Lyso-Gb3 biomarker levels are normal in D313Y carriers.2 ClinVar reports this variant as Benign by 10 clinical laboratories and Likely benign by 7, with only 4 reporting Uncertain significance and 2 reporting other. The majority consensus supports a benign interpretation.3 Multiple in silico predictors support a benign interpretation: SpliceAI predicts no splicing impact (max delta = 0.00), and BayesDel score of 0.065 is in the benign range.4 Based on the generic ACMG/AMP 2015 framework, the combined evidence includes BS1 (supporting benign), BS2 (supporting benign), BS3 (strong benign), BP4 (supporting benign), and BP6 (supporting benign). No pathogenic criteria are met. The overall classification is Benign.5
GLA
Final classification
Likely Benign
GLA c.937G>T · p.Asp313Tyr
GLA
NM_000169.3:c.937G>T (p.Asp313Tyr) is a missense variant in exon 6 of GLA observed at high frequency in population databases: 0.304% in gnomAD v2.1 (624/205,260 alleles, 3 homozygotes) and 0.378% in gnomAD v4.1 (4,565/1,207,947 alleles, 10 homozygotes).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 supporting, BS2 supporting, BS3 strong, BP4 supporting, BP6 supporting; combination = 1 strong benign + 4 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BS2BS3BP4BP6
Likely Benign
GLA c.937G>T
BS1 + BS2 + BS3 + BP4 + BP6
→
Likely Benign
4
spliceai ↗bayesdel
5
generic_acmg_combination_rules
Gene diagram
· NM_000169.3 · variants mapped to exon structure
GLA
NM_000169.3
Fetching transcript structure from UCSC…
Applied criteria · 5 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign

gnomAD v4.1
gnomAD v2.1
v4.1
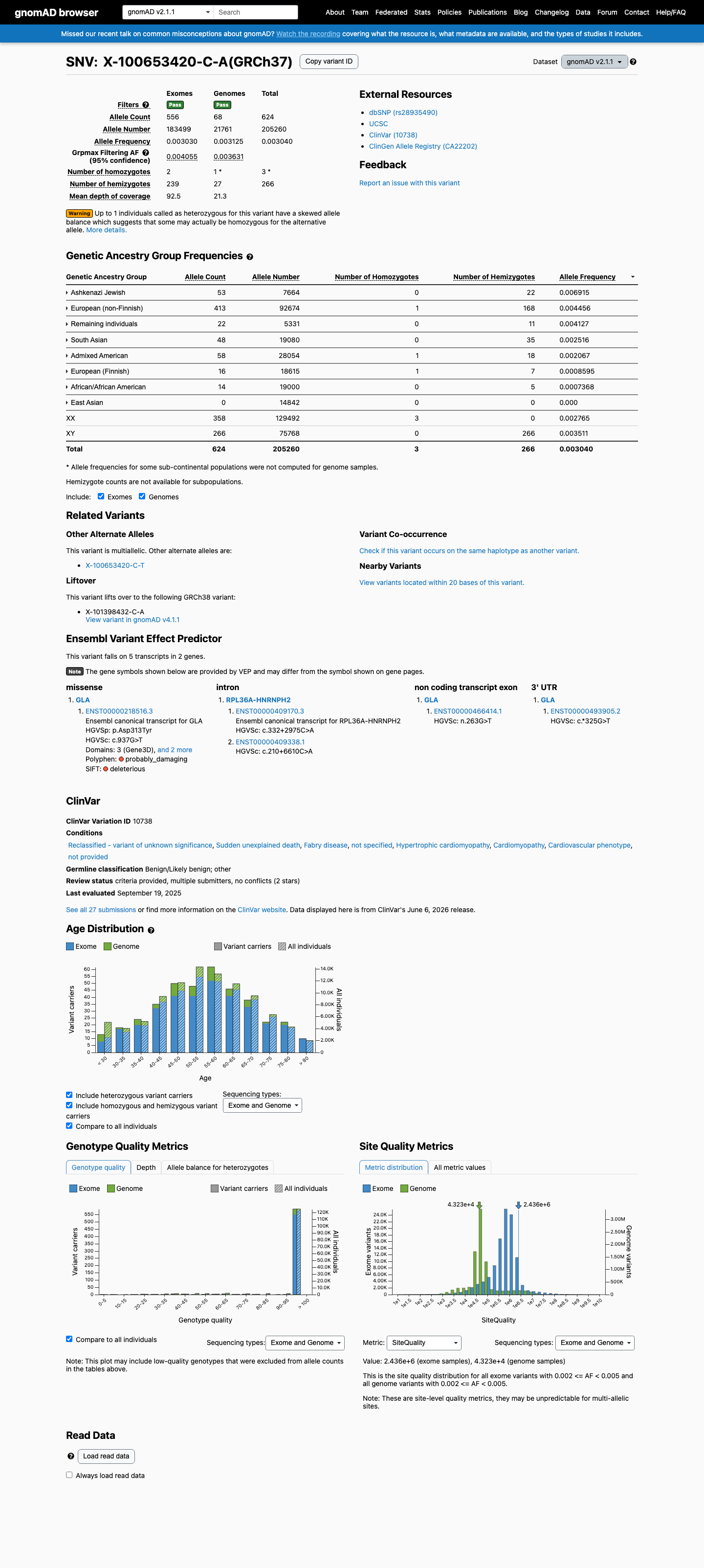
This variant is present in gnomAD v4.1 (AF= 0.00377914; MAF= 0.37791%, 4565/1207947 alleles, homozygotes = 10) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00626703; MAF= 0.62670%, 138/22020 alleles, homozygotes = 1); grpmax FAF= 0.0042627.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00304005; MAF= 0.30400%, 624/205260 alleles, homozygotes = 3) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00691545; MAF= 0.69154%, 53/7664 alleles, homozygotes = 0); grpmax FAF= 0.0040545.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0029704338214976513, 43/14476 alleles, homozygotes = 1).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.38%
· 4565 / 1,207,947
10 hom · FAF 0.43%
10 hom · FAF 0.43%
Ashkenazi Jewish 138 / 22,020 |
0.63% 1 hom |
European (non-Finnish) 3912 / 893,709 |
0.44% 5 hom |
Middle Eastern 18 / 4,329 |
0.42% |
Remaining individuals 157 / 47,550 |
0.33% 1 hom |
South Asian 183 / 56,710 |
0.32% 2 hom |
Admixed American 88 / 45,679 |
0.19% 1 hom |
European (Finnish) 35 / 46,477 |
0.075% |
African/African American 34 / 57,057 |
0.06% |
+ 2 not observed (Amish, East Asian)
gnomAD v2.1
0.3%
· 624 / 205,260
3 hom · FAF 0.41%
3 hom · FAF 0.41%
Ashkenazi Jewish 53 / 7,664 |
0.69% |
European (non-Finnish) 413 / 92,674 |
0.45% 1 hom |
Remaining individuals 22 / 5,331 |
0.41% |
South Asian 48 / 19,080 |
0.25% |
Admixed American 58 / 28,054 |
0.21% 1 hom |
European (Finnish) 16 / 18,615 |
0.086% 1 hom |
African/African American 14 / 19,000 |
0.074% |
+ 1 not observed (East Asian)
gnomAD Canada 🇨🇦
0.3%
· 43 / 14,476
1 hom · FAF 0.2%
1 hom · FAF 0.2%
indel · split
Ashkenazi Jewish 4 / 625 |
0.64% 1 hom |
South Asian 5 / 993 |
0.5% |
European (non-Finnish) 31 / 9,401 |
0.33% |
Latino/Admixed American 2 / 646 |
0.31% |
Remaining individuals 1 / 870 |
0.11% |
+ 4 not observed (African/African American, East Asian, European (Finnish), Middle Eastern)
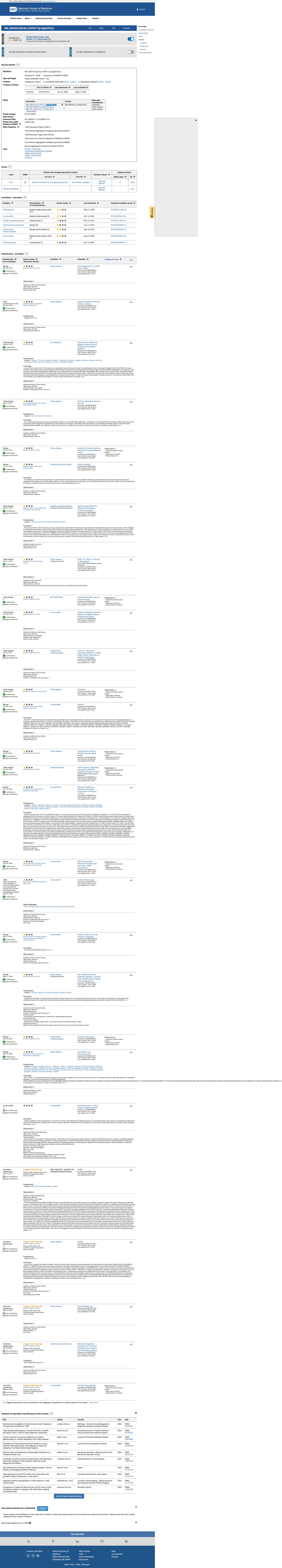
ClinVar
This variant has been reported in ClinVar as Benign (10 clinical laboratories) and as Likely benign (7 clinical laboratories) and as Uncertain significance (4 clinical laboratories) and as other (2 clinical laboratories). (ClinVarID = 10738)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). BayesDel score = 0.0650709.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV54510729, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Fabry disease: characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele.
Searched
c.937G>TD313YAsp313Tyr
Found
D313Y was characterized as a pseudodeficiency allele. Expressed D313Y retained ~60% WT enzymatic activity in COS-7 cells, localized properly to lysosomes, and was stable at lysosomal pH 4.6 but showed reduced activity at neutral pH 7.4. D313Y occurred in 0.45% of 883 normal X-chromosomes. The D313Y substitution is structurally tolerated: Asp313 is solvent-exposed in a peripheral alpha-helix, not conserved in evolution, and the tyrosine substitution is easily accommodated. The authors conclude D313Y is a rare coding sequence variant causing pseudodeficiency in plasma.
Variant
✓ Names this variant — characterised directly
Applied to
→BS3 supports · met
Why
Key functional evidence establishing D313Y as a pseudodeficiency allele with ~60% WT activity and normal lysosomal localization; referenced in BS3 assessment as strong benign evidence.
Thus, D313Y is a rare exonic variant with about 60% of wild-type activity in vitro and reduced activity at neutral pH, resulting in low plasma a-Gal A activity.
Location Abstract; Results (Mutation Detection and Frequency, Expression Studies, pH Stability); Discussion; Figure 1-4 · Context COS-7 cell transient transfection; alpha-galactosidase A fluorogenic enzyme assay; immunofluorescence microscopy with LAMP-2 (lysosomal) and BiP (ER) markers; pH stability assay; molecular homology modeling based on chicken alpha-Gal B crystal structure · full text
Lyso-Gb3 Indicates that the Alpha-Galactosidase A Mutation D313Y is not Clinically Relevant for Fabry Disease.
Searched
D313YLyso-Gb3
Found
Lyso-Gb3 biomarker analysis in D313Y carriers demonstrated normal levels, indicating absence of glycolipid accumulation characteristic of Fabry disease. The authors conclude that D313Y is not clinically relevant for Fabry disease and that reduced alpha-galactosidase A activity in plasma represents a pseudodeficiency rather than true enzyme deficiency.
Variant
✓ Names this variant — characterised directly
Applied to
→BS3 supports · met
Why
Key evidence confirming D313Y is a pseudodeficiency allele without biochemical evidence of Fabry disease; referenced in BS3 assessment as strong benign evidence.
Lyso-Gb3 Indicates that the Alpha-Galactosidase A Mutation D313Y is not Clinically Relevant for Fabry Disease.
Location Abstract; Results; Discussion · full text
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
19085643 ↗
[Ophthalmological manifestations in Fabry's disease. Four clinical cases showing deficient alpha-galactosidase-A activity].
CLINVAR
20110537 ↗
Mutations of the GLA gene in young patients with stroke: the PORTYSTROKE study--screening genetic conditions in Portuguese young stroke patients.
CLINVAR
23219219 ↗
Phenotypical characterization of α-galactosidase A gene mutations identified in a large Fabry disease screening program in stroke in the young.
CLINVAR
23393592 ↗
Multifocal white matter lesions associated with the D313Y mutation of the α-galactosidase A gene.
CLINVAR