NM_000179.2:c.188C>T (p.Ser63Phe) is a missense variant in MSH6 exon 1. The variant is extremely rare, observed in 1 of 1,509,420 alleles (AF = 6.625e-07) in gnomAD v4.1 and absent from gnomAD v2.1, meeting PM2_Supporting under the InSiGHT MSH6 VCEP v2.0.0 framework.1 Multiple in silico predictors are consistent with a benign effect. The HCI prior probability for p.Ser63Phe is 0.003, meeting BP4_Supporting (threshold < 0.11). REVEL score is 0.195, BayesDel score is -0.269, and SpliceAI predicts no splicing impact (max delta = 0.00).2 PVS1 is not applicable (missense variant). PS1 is not met (no alternate nucleotide change encoding p.Ser63Phe classified as P/LP by this VCEP). PM5 is not met because PP3 is not supporting (HCI prior 0.003, far below PP3_Supporting threshold of >0.68). Multiple VCEP criteria are explicitly Not Applicable: PS4, PP5, PM1, PM6, PP2, BP1, BP2, BP6.3 The variant has been reported in ClinVar as Uncertain significance by two clinical laboratories (VariationID: 1053294). It has been observed in somatic cancers (COSMIC COSV52275784, n = 6). No variant-specific functional data, segregation data, de novo occurrences, or tumor phenotype data were identified.4 At present, the only scored criteria are PM2_Supporting (pathogenic) and BP4_Supporting (benign), which offset. Multiple criteria remain unassessed due to absence of clinical, functional, and segregation data. Comprehensive assessment awaits functional assay results for p.Ser63Phe, tumor MSI/IHC data from variant carriers, and segregation analysis.
MSH6
Final classification
Uncertain Significance - Conflicting Evidence
MSH6 c.188C>T · p.Ser63Phe
MSH6
NM_000179.2:c.188C>T (p.Ser63Phe) is a missense variant in MSH6 exon 1. The variant is extremely rare, observed in 1 of 1,509,420 alleles (AF = 6.625e-07) in gnomAD v4.1 and absent from gnomAD v2.1, meeting PM2_Supporting under the InSiGHT MSH6 VCEP v2.0.0 framework.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MSH6 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule31 (Benign.Supporting >=1 + Pathogenic.Supporting >=1) with applied criteria: PM2 supporting, BP4 supporting benign; maps to Uncertain Significance - Conflicting Evidence.
Classification rationale
PM2
BP4
Uncertain Significance - Conflicting Evidence
MSH6 c.188C>T
PM2 + BP4
→
Uncertain Significance - Conflicting Evidence
2
hci_priorrevelbayesdelspliceai ↗cspec ↗
3
cspec ↗hci_prior
Gene diagram
· NM_000179.2 · variants mapped to exon structure
MSH6
NM_000179.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 14 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000179.2:c.188C>T is extremely rare in gnomAD v4.1 (1/1,509,420 alleles; AF = 6.625e-07), meeting the MSH6 VCEP PM2_Supporting threshold of allele frequency < 0.00002 (fewer than 1 in 50,000 alleles). The variant is absent from gnomAD v2.1 and absent from all populations except a single European (non-Finnish) allele.
gnomAD v4.1: 1/1509420 total alleles (AF = 6.625e-07)
✓
BP4
supporting
Benign
The HCI prior probability for p.Ser63Phe is 0.003, meeting the MSH6 VCEP BP4_Supporting threshold of < 0.11. Multiple in silico predictors are consistent with a benign effect: REVEL score is 0.195 (below typical pathogenic threshold), BayesDel score is -0.269 (negative, favoring benign), and SpliceAI predicts no splicing impact (max delta = 0.00).
HCI prior = 0.003 (threshold BP4_Supporting: < 0.11met). REVEL = 0.195. BayesDel = -0.269. SpliceAI max delta = 0.00. Multiple in silico tools converge on a benign prediction.
Assessed · not applied
Pathogenic
PS1
No alternative nucleotide change encoding p.Ser63Phe has been classified by the InSiGHT Hereditary Colorectal Cancer/Polyposis EP as Pathogenic or Likely Pathogenic.
PS2
No de novo occurrence with confirmed parentage has been identified for NM_000179.2:c.188C>T in the available literature, ClinVar submissions, or databases.
PS3
No calibrated functional assay data for p.Ser63Phe was identified in the VCEP functional assay documentation spreadsheet or in available literature.
PM5
The MSH6 VCEP PM5 rule requires PP3 to be supporting for the missense change as a prerequisite.
PP1
No co-segregation data with disease in pedigrees has been identified for NM_000179.2:c.188C>T.
PP3
The HCI prior probability for p.Ser63Phe is 0.003, which falls far below the MSH6 VCEP PP3_Supporting threshold of >0.68.
PP4
No tumor MSI/IHC data for patients harboring NM_000179.2:c.188C>T was identified in the available evidence.
Benign
BA1
The MSH6 VCEP BA1 threshold requires gnomAD v4 Grpmax filtering allele frequency ≥ 0.0022 (0.22%).
BS1
The MSH6 VCEP BS1 threshold requires gnomAD v4 Grpmax filtering allele frequency ≥ 0.00022 (0.022%).
BS2
No evidence of co-occurrence in trans with a known pathogenic MSH6 variant in a patient with colorectal cancer after age 45 without CMMRD features has been identified.
BS3
No calibrated functional assay data demonstrating proficient MMR function for p.Ser63Phe is available.
BS4
No lack-of-segregation data (variant present in unaffected family members) has been identified for NM_000179.2:c.188C>T.
BP5
No tumor data (CRC/Endometrial MSS status, MMR protein expression, BRAF V600E, or MLH1 methylation) is available for patients harboring NM_000179.2:c.188C>T.
BP7
NM_000179.2:c.188C>T is a missense variant (p.Ser63Phe), not a synonymous (silent) or intronic variant at or beyond -21/+7.
N/A · 9
PVS1 · PS4 · PM1 · PM6 · PP2 · PP5 · BP1 · BP2 · BP6
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
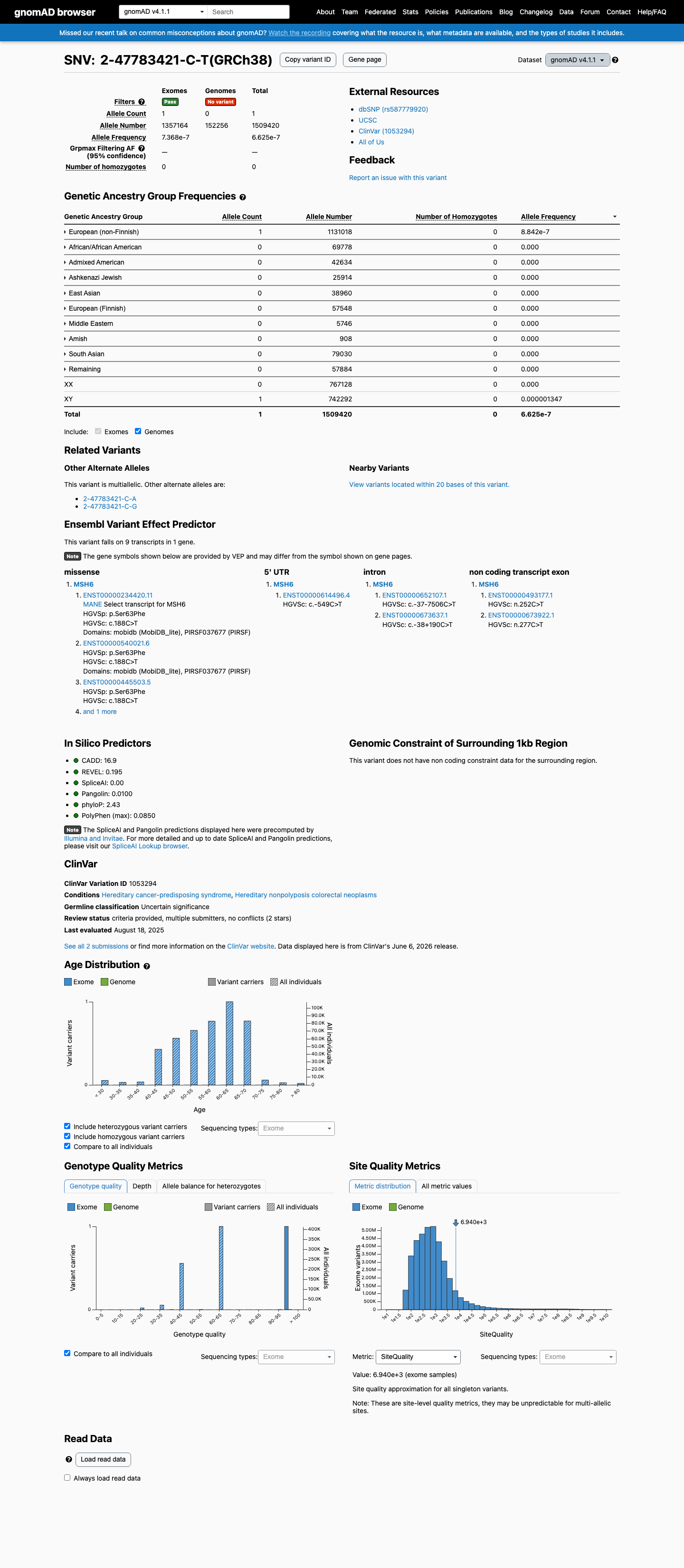
This variant is present in gnomAD v4.1 (AF= 6.62506e-07; MAF= 0.00007%, 1/1509420 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.84159e-07; MAF= 0.00009%, 1/1131018 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Not available in gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.6e-05%
· 1 / 1,509,420
0 hom
0 hom
European (non-Finnish) 1 / 1,131,018 |
8.8e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
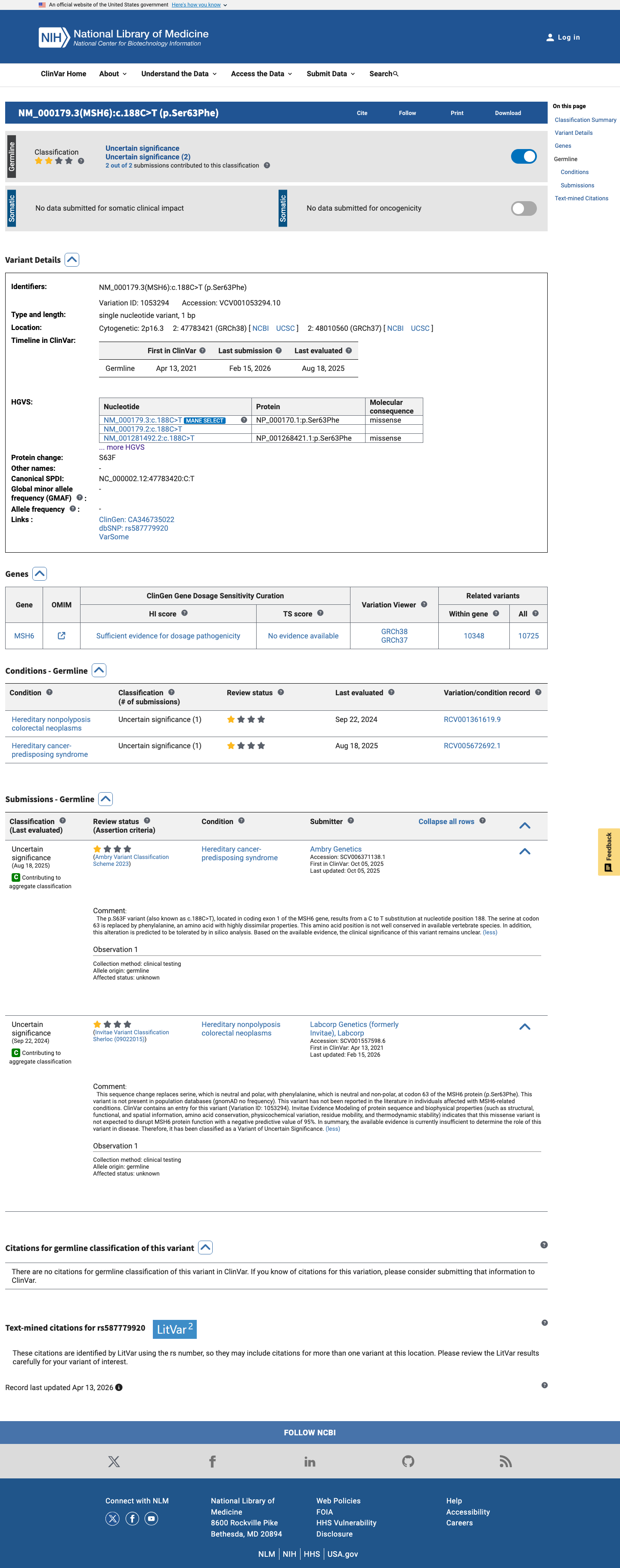
This variant has been reported in ClinVar as Uncertain significance (2 clinical laboratories). (ClinVarID = 1053294)
In silico
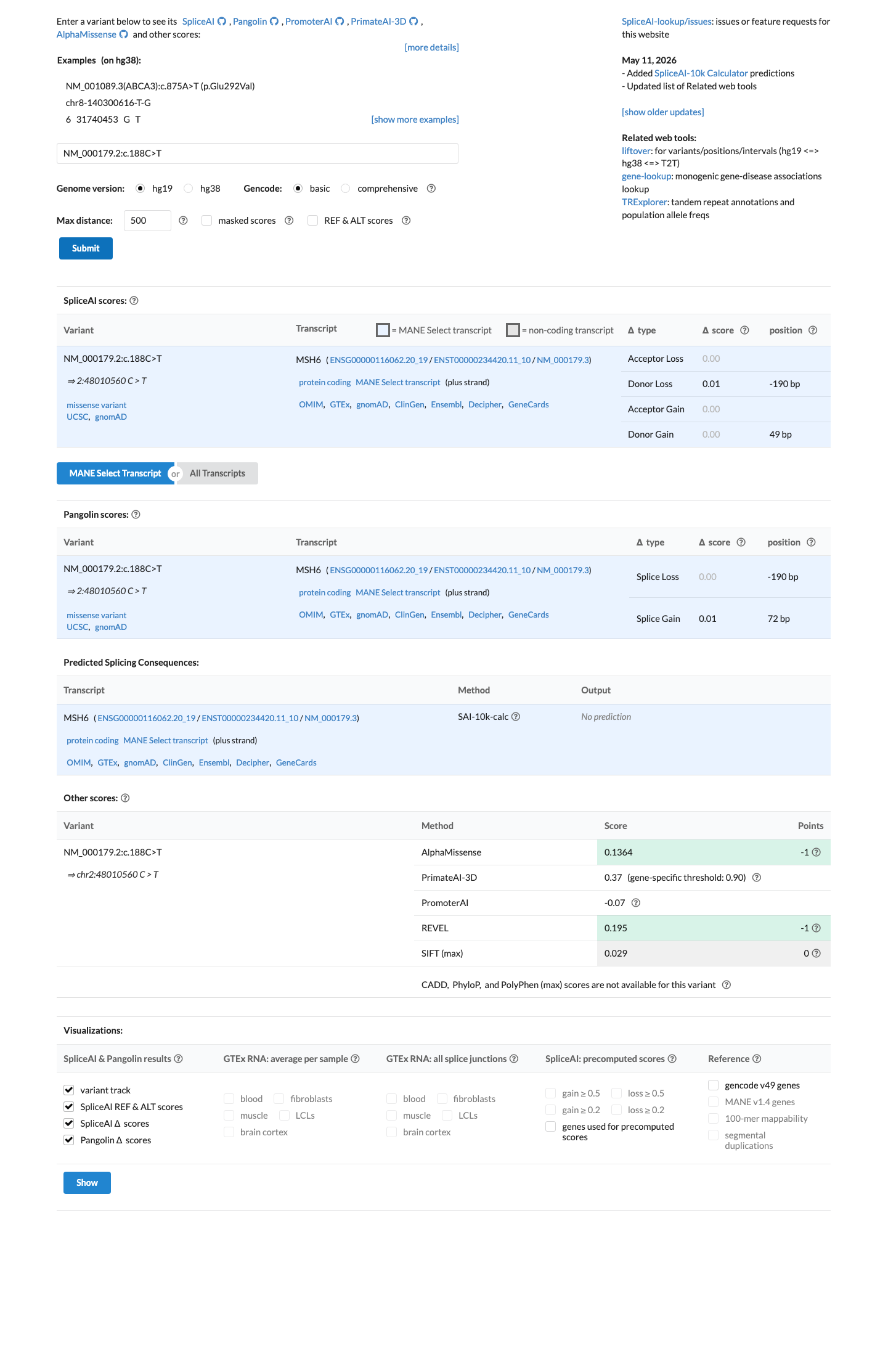
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.195. BayesDel score = -0.269383. HCI prior probability for pathogenicity = 0.003. Custom PP2 score = 0.027.
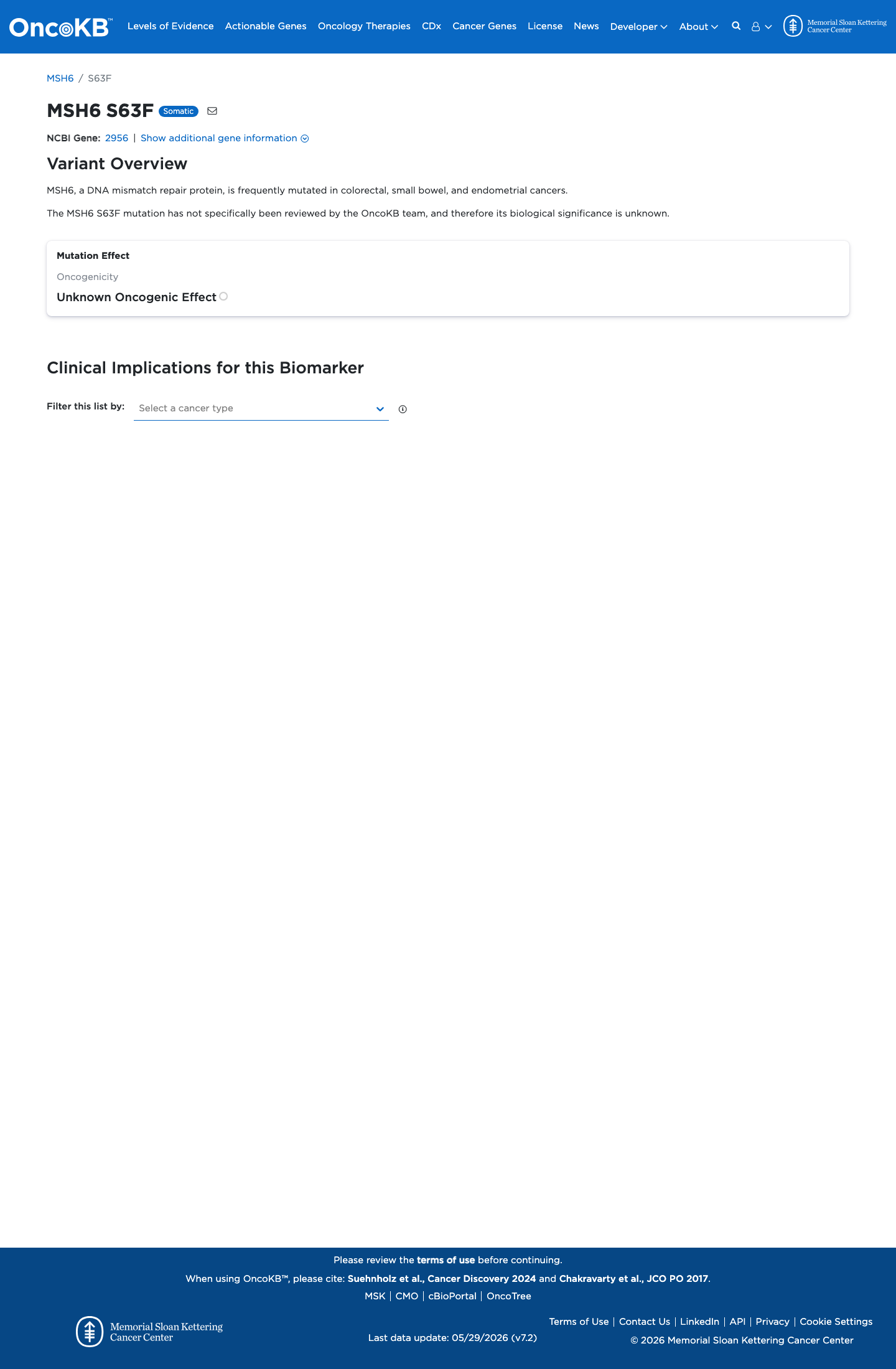
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MSH6, a DNA mismatch repair protein, is frequently mutated in colorectal, small bowel, and endometrial cancers.
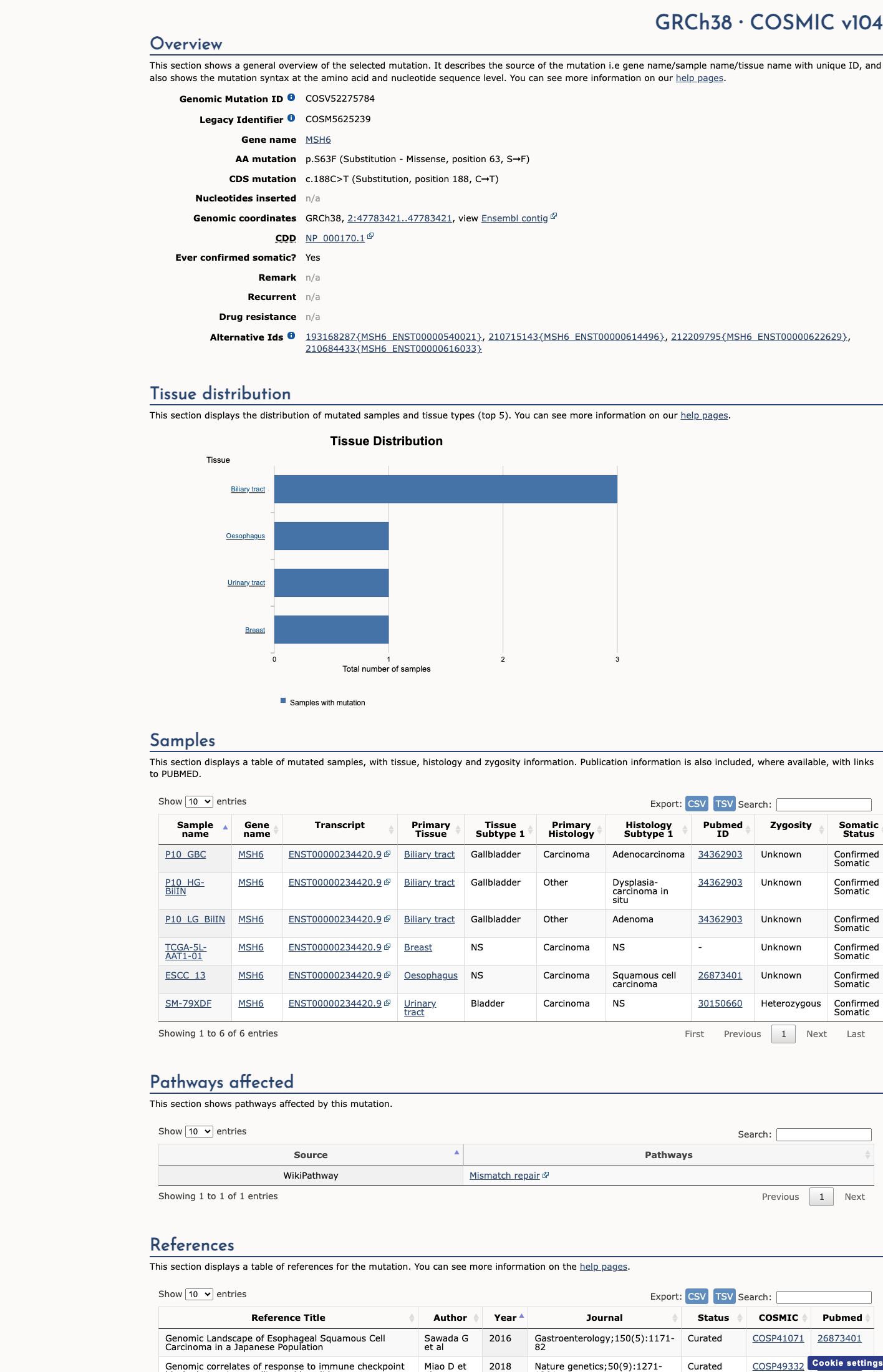
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV52275784, n = 6 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 2 PMIDs not cited in assessment
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR