Classification rationale
PM2PVS1
VUS
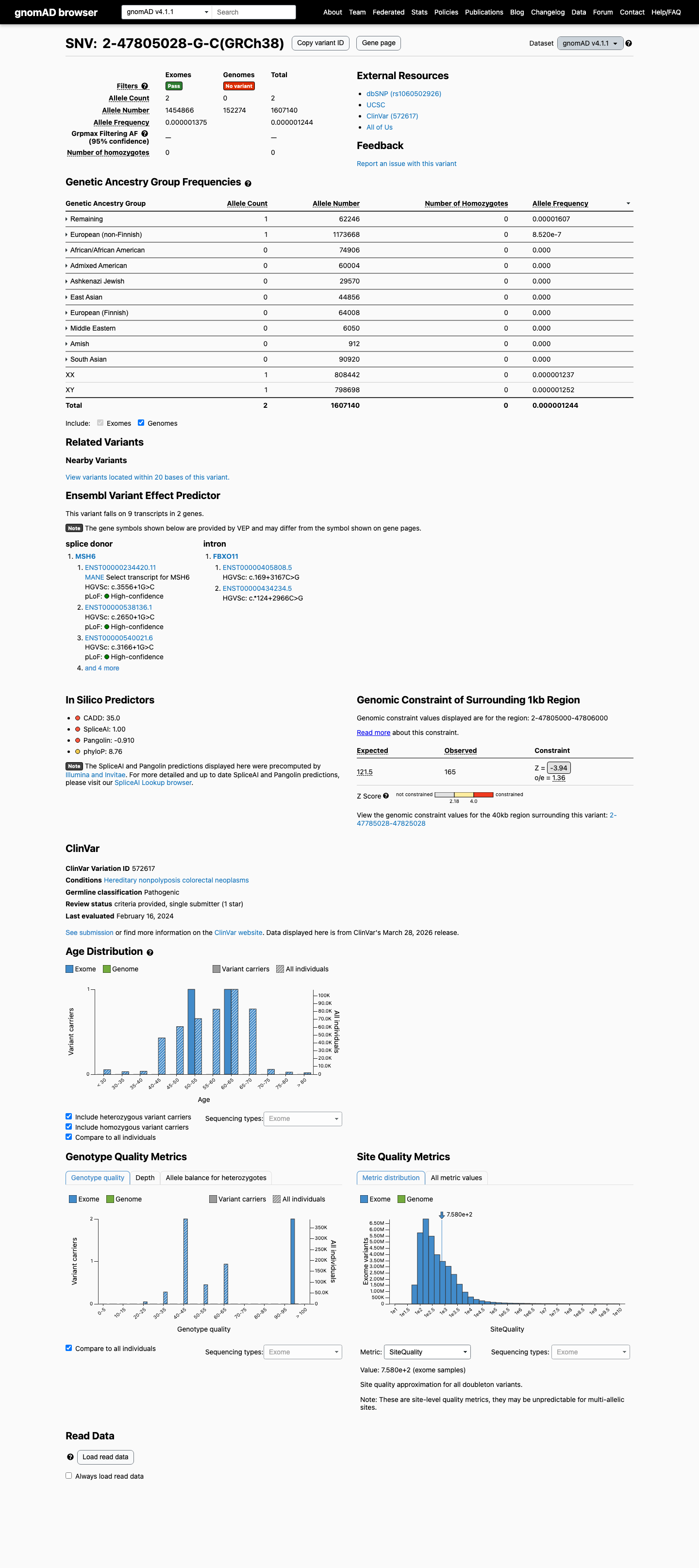
MSH6 c.3556+1G>C
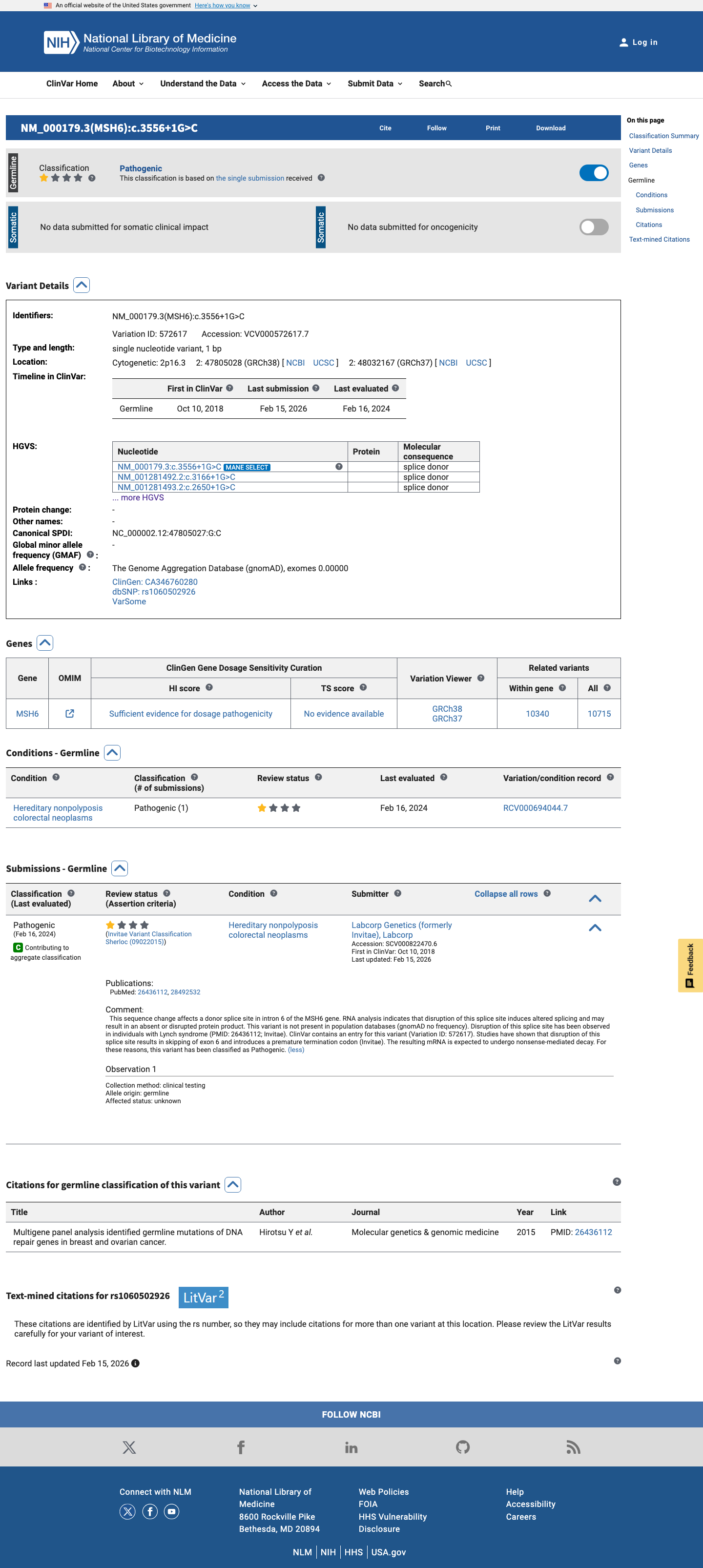
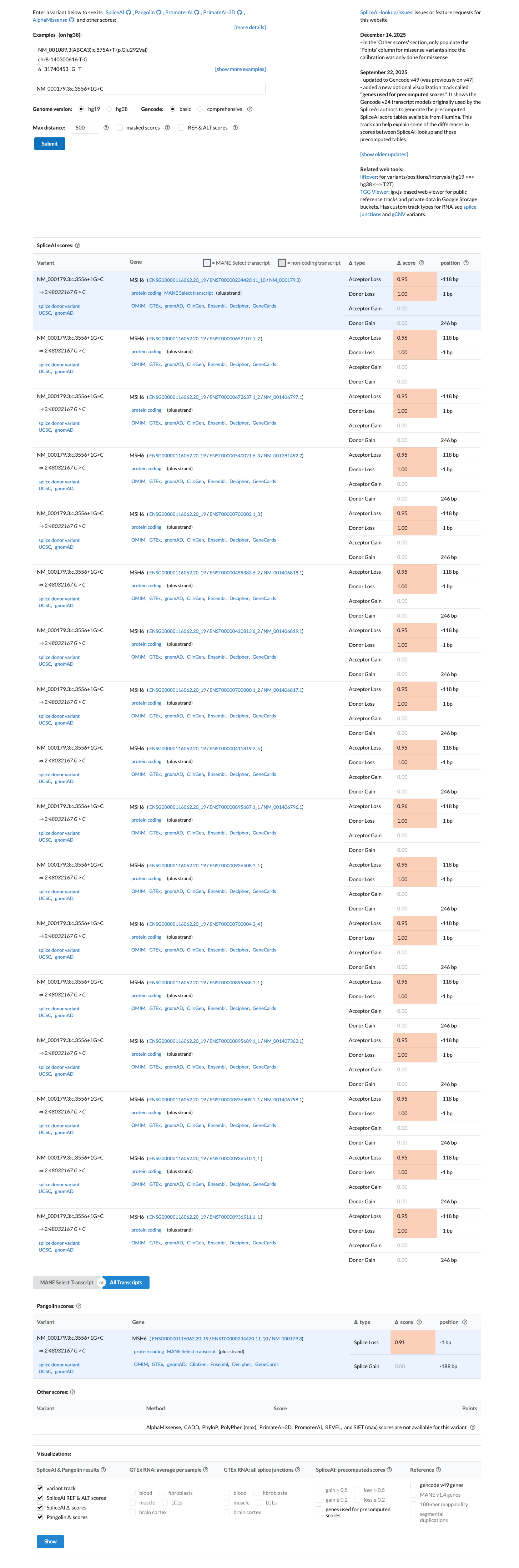
The MSH6 NM_000179.3:c.3556+1G>C (NP_000170.1:p.?) variant has been reported in ClinVar as Pathogenic by a single clinical laboratory.1 This variant is absent from gnomAD v2.1 and is present in gnomAD v4.1 at a total allele frequency of 1.24445e-06 (2/1,607,140 alleles), which is below the MSH6 VCEP PM2 threshold of 0.00002.2 This canonical +1 splice-donor variant is predicted to disrupt splicing, with SpliceAI showing a maximum delta score of 1.00, and the MSH6 VCEP PVS1 framework supports very strong pathogenic evidence for canonical splice variants expected to cause a frameshifting transcript subject to nonsense-mediated decay.3
PM2 + PVS1
→
VUS
3
spliceai ↗cspec ↗pvs1_gene_contextpvs1_variant_assessment