NM_000179.3:c.4002-26_4002-25insCT is present in gnomAD v4.1 at a grpmax filtering allele frequency of 2.41% in the East Asian population, exceeding the InSiGHT MSH6 VCEP BA1 stand-alone benign threshold of 0.22%. This population frequency is incompatible with a pathogenic role in Lynch syndrome.1 SpliceAI predicts no splicing impact (max delta = 0.02), meeting the VCEP BP4_Supporting criterion for intronic variants.2 The variant is located at intronic positions -26/-25, satisfying the VCEP BP7_Supporting criterion for intronic variants at or beyond -21/+7.3 No pathogenic evidence criteria are met: PVS1 is not applicable (deep intronic, no null-allele evidence); PM2 is not met (gnomAD v4.1 AF = 0.185%); PP3 is not met (SpliceAI delta 0.02 < 0.2); and no functional, segregation, de novo, or tumor phenotype data support pathogenicity.4
MSH6
Final classification
Benign
MSH6 c.4002-26_4002-25insCT · p.?
MSH6
NM_000179.3:c.4002-26_4002-25insCT is present in gnomAD v4.1 at a grpmax filtering allele frequency of 2.41% in the East Asian population, exceeding the InSiGHT MSH6 VCEP BA1 stand-alone benign threshold of 0.22%. This population frequency is incompatible with a pathogenic role in Lynch syndrome.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MSH6 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule17 (1 Benign.Stand Alone) with applied criteria: BA1 stand-alone benign, BP4 supporting benign, BP7 supporting benign; maps to Benign.
Classification rationale
BA1BP4BP7
Benign
MSH6 c.4002-26_4002-25insCT
BA1 + BP4 + BP7
→
Benign
Gene diagram
· NM_000179.3 · variants mapped to exon structure
MSH6
NM_000179.3
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 13 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
The grpmax filtering allele frequency in gnomAD v4.1 is 0.0241 (2.41%) in the East Asian population, which exceeds the VCEP BA1 threshold of ≥0.0022 (0.22%). The variant has a total v4.1 allele frequency of 0.00185 (186/100,732 alleles) and is not known to be a founder pathogenic variant. This frequency is incompatible with MSH6-associated Lynch syndrome prevalence.
gnomAD v4.1 grpmax FAF = 0.0240982 (2.41%) in East Asian population.gnomAD v4.1 total AF = 0.00184648 (186/100732 alleles
✓
BP4
supporting
Benign
For intronic variants, the VCEP BP4_Supporting rule is met when SpliceAI predicts no splicing impact with a delta score ≤ 0.1. SpliceAI predicts no significant splice impact for this variant (max delta = 0.02).
SpliceAI max delta score = 0.02≤ 0.1 threshold for BP4_Supporting.
✓
BP7
supporting
Benign
NM_000179.3:c.4002-26_4002-25insCT is an intronic variant located at positions -26 and -25 relative to the exon 10 boundary, which is beyond -21 from the splice acceptor site. This satisfies the VCEP BP7 rule for intronic variants at or beyond -21/+7.
Variant is an intronic insertion at c.4002-26_4002-25 (intron 9)located beyond the -21 intronic boundary specified by the VCEP BP7 rule.
Assessed · not applied
Pathogenic
PVS1
NM_000179.3:c.4002-26_4002-25insCT is a deep intronic 2-bp insertion in intron 9, 26-25 nucleotides upstream of exon 10.
PS1
PS1 requires either the same amino acid change as a known pathogenic missense variant, or a variant at the same non-canonical splice nucleotide as a confirmed pathogenic splice variant with similar or worse SpliceAI prediction.
PS2
No de novo observations of NM_000179.3:c.4002-26_4002-25insCT have been reported in ClinVar, the published literature, or public databases.
PS3
No functional studies (MMR activity assays, protein expression, or splicing reporter assays) directly testing NM_000179.3:c.4002-26_4002-25insCT were identified.
PM2
The VCEP PM2 (Supporting) threshold requires absent or extremely rare allele frequency (<0.00002; <1 in 50,000 alleles) in gnomAD v4.
PP1
No cosegregation data are available for NM_000179.3:c.4002-26_4002-25insCT.
PP3
For intronic variants, the VCEP PP3_Supporting rule requires SpliceAI delta score ≥ 0.2 for a predicted splice defect at non-canonical nucleotides.
PP4
No tumor MSI or immunohistochemistry data are available for patients carrying NM_000179.3:c.4002-26_4002-25insCT.
Benign
BS1
The VCEP BS1 (Strong) threshold is grpmax FAF ≥ 0.00022 and < 0.0022 (0.022-0.22%).
BS2
BS2 requires co-occurrence in trans with a known pathogenic MSH6 variant in a patient with colorectal cancer after age 45 without CMMRD features.
BS3
No calibrated functional assay data or laboratory-based mRNA splicing assays are available for NM_000179.3:c.4002-26_4002-25insCT.
BS4
No pedigrees demonstrating lack of cosegregation with disease have been reported for this variant.
BP5
BP5 requires MSS colorectal/endometrial tumors or BRAF V600E/MLH1 methylation with MSI-H/MLH1 loss.
N/A · 11
PS4 · PM1 · PM4 · PM5 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6
Research & evidence
Population frequency · supports benign

gnomAD v4.1
gnomAD v2.1
v4.1
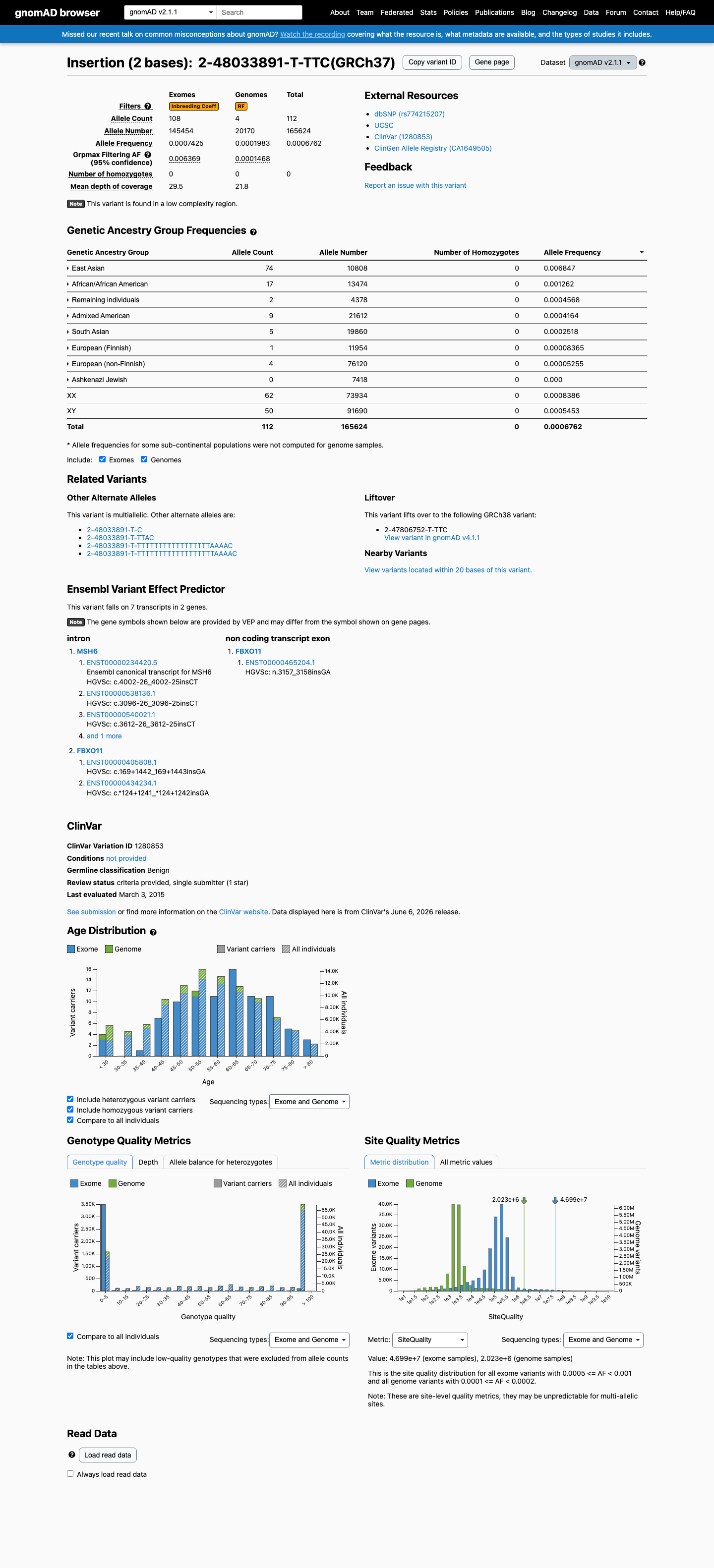
This variant is present in gnomAD v4.1 (AF= 0.00184648; MAF= 0.18465%, 186/100732 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.0285171; MAF= 2.85171%, 105/3682 alleles, homozygotes = 0); grpmax FAF= 0.0240982.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00067623; MAF= 0.06762%, 112/165624 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.00684678; MAF= 0.68468%, 74/10808 alleles, homozygotes = 0); grpmax FAF= 0.00636889.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.18%
· 186 / 100,732
0 hom · FAF 2.4%
0 hom · FAF 2.4%
East Asian 105 / 3,682 |
2.9% |
Middle Eastern 2 / 292 |
0.68% |
Remaining individuals 15 / 3,058 |
0.49% |
South Asian 14 / 5,426 |
0.26% |
Admixed American 9 / 8,196 |
0.11% |
African/African American 9 / 10,398 |
0.087% |
European (non-Finnish) 31 / 62,992 |
0.049% |
European (Finnish) 1 / 4,598 |
0.022% |
+ 2 not observed (Amish, Ashkenazi Jewish)
gnomAD v2.1
0.068%
· 112 / 165,624
0 hom · FAF 0.64%
0 hom · FAF 0.64%
East Asian 74 / 10,808 |
0.68% |
African/African American 17 / 13,474 |
0.13% |
Remaining individuals 2 / 4,378 |
0.046% |
Admixed American 9 / 21,612 |
0.042% |
South Asian 5 / 19,860 |
0.025% |
European (Finnish) 1 / 11,954 |
0.0084% |
European (non-Finnish) 4 / 76,120 |
0.0053% |
+ 1 not observed (Ashkenazi Jewish)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Benign (1 clinical laboratory). (ClinVarID = 1280853)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links