NM_000222.2:c.1990+8C>T is an intronic variant at position +8 in intron 13 of KIT, classified as Likely benign based on the generic ACMG/AMP 2015 framework (PMID:25741868).1 SpliceAI predicts no significant splicing impact (max delta score=0.02), meeting BP4 at supporting benign level.2 The variant is classified as Likely benign in ClinVar (VC ID 415791) by Labcorp Genetics/Invitae, meeting BP6 at supporting benign level.3 The variant is present at extremely low frequency in gnomAD (v2.1 AF=0.00279%, v4.1 AF=0.00178%), meeting PM2 at supporting pathogenic level.4 With two supporting benign criteria (BP4, BP6) and one supporting pathogenic criterion (PM2), the net evidence meets the generic ACMG/AMP threshold for Likely benign (≥2 supporting benign criteria).5
KIT
Final classification
Likely Benign
KIT c.1990+8C>T · p.?
KIT
NM_000222.2:c.1990+8C>T is an intronic variant at position +8 in intron 13 of KIT, classified as Likely benign based on the generic ACMG/AMP 2015 framework (PMID:25741868).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign, BP6 supporting benign; combination = 1 supporting + 2 supporting benign, which maps to Likely Benign.
Classification rationale
PM2
BP4BP6
Likely Benign
KIT c.1990+8C>T
PM2 + BP4 + BP6
→
Likely Benign
1
generic_acmg_combination_rules
5
generic_acmg_combination_rules
Gene diagram
· NM_000222.2 · variants mapped to exon structure
KIT
NM_000222.2
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 15 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in population databases (gnomAD v2.1 AF=0.00279%, v4.1 AF=0.00178%), well below the 0.1% non-VCEP PM2 threshold. One homozygote is observed in each gnomAD version (South Asian subpopulation), which is noted but does not disqualify PM2 at this allele frequency.
gnomAD v2.1: 7/250542 alleles (AF=0.00279%)gnomAD v4.1: 28/1
✓
BP4
supporting
Benign
SpliceAI predicts no significant splicing impact for this intronic variant (max delta score=0.02). While REVEL and BayesDel are unavailable for intronic variants, SpliceAI is the primary relevant in silico tool for splice region variants and supports a benign interpretation.
SpliceAI max delta=0.02 (no splice impact predicted)
✓
BP6
supporting
Benign
This variant is classified as Likely benign in ClinVar (VC ID 415791) by a clinical testing laboratory (Labcorp Genetics/Invitae), which constitutes a reputable source reporting the variant as benign.
ClinVar classification: Likely benignsubmitter: Labcorp Genetics (formerly Invitae)review status: criteria provided
Assessed · not applied
Pathogenic
PS2
No de novo data with confirmed maternity and paternity were identified in ClinVar submissions or the literature for this variant.
PS3
No well-established functional studies were identified for this variant.
PS4
No case-control or cohort data demonstrating enrichment of this variant in affected individuals were identified.
PM6
PM6 requires a de novo observation without confirmation of maternity and paternity.
PP1
No cosegregation data in affected families were identified for this variant.
PP3
Computational evidence does not support a deleterious effect.
PP4
No patient phenotype or family history data were provided for this variant.
PP5
PP5 requires a reputable source reporting the variant as pathogenic.
Benign
BA1
BA1 requires allele frequency >1% in any population.
BS1
BS1 threshold for non-VCEP is AF >0.3%.
BS2
A homozygous individual is observed in gnomAD (South Asian subpopulation), which is notable for an autosomal dominant KIT-related disorder.
BS3
No well-established functional studies demonstrating no damaging effect were identified for this variant.
BS4
No segregation data in affected families are available for this variant.
BP2
No data on cis/trans phasing with pathogenic variants are available for this variant.
BP5
BP5 requires observation of the variant in a case with an alternate molecular basis for disease.
N/A · 10
PVS1 · PS1 · PM1 · PM3 · PM4 · PM5 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports benign
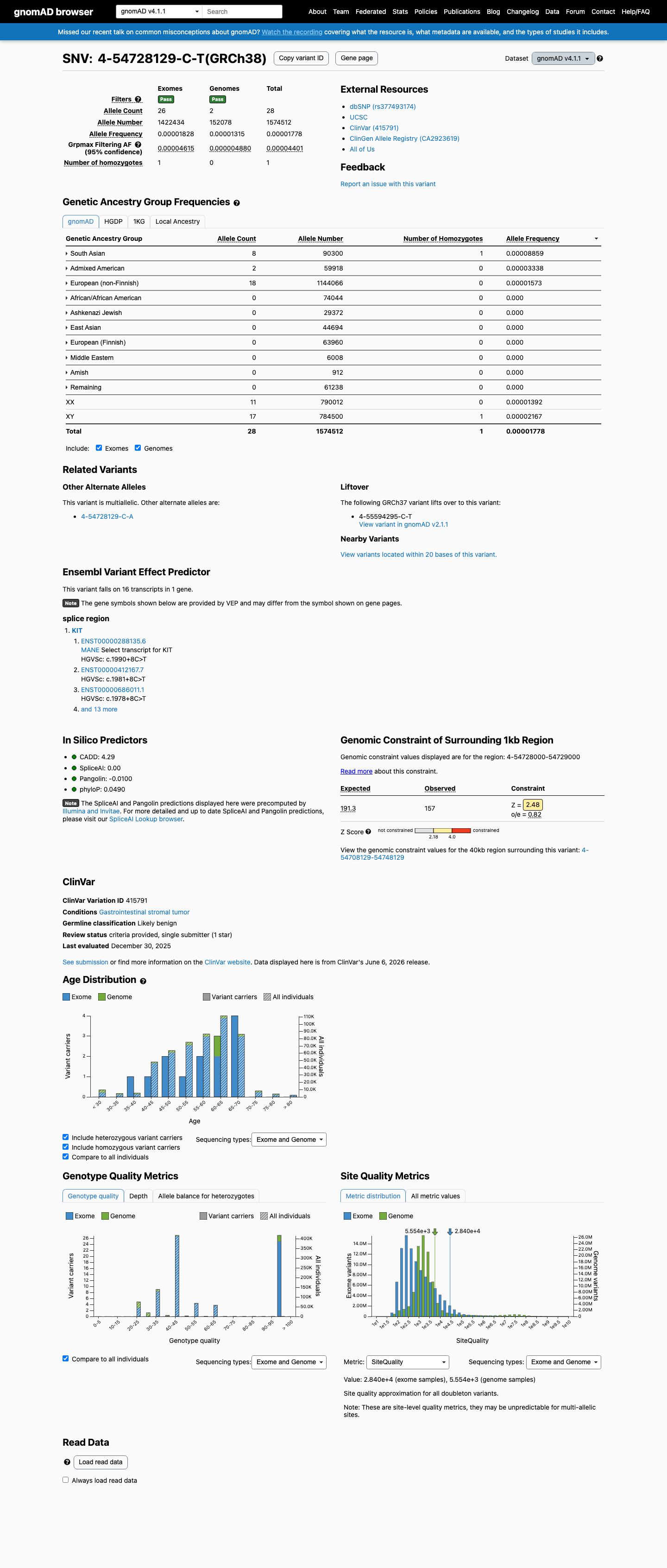
gnomAD v4.1
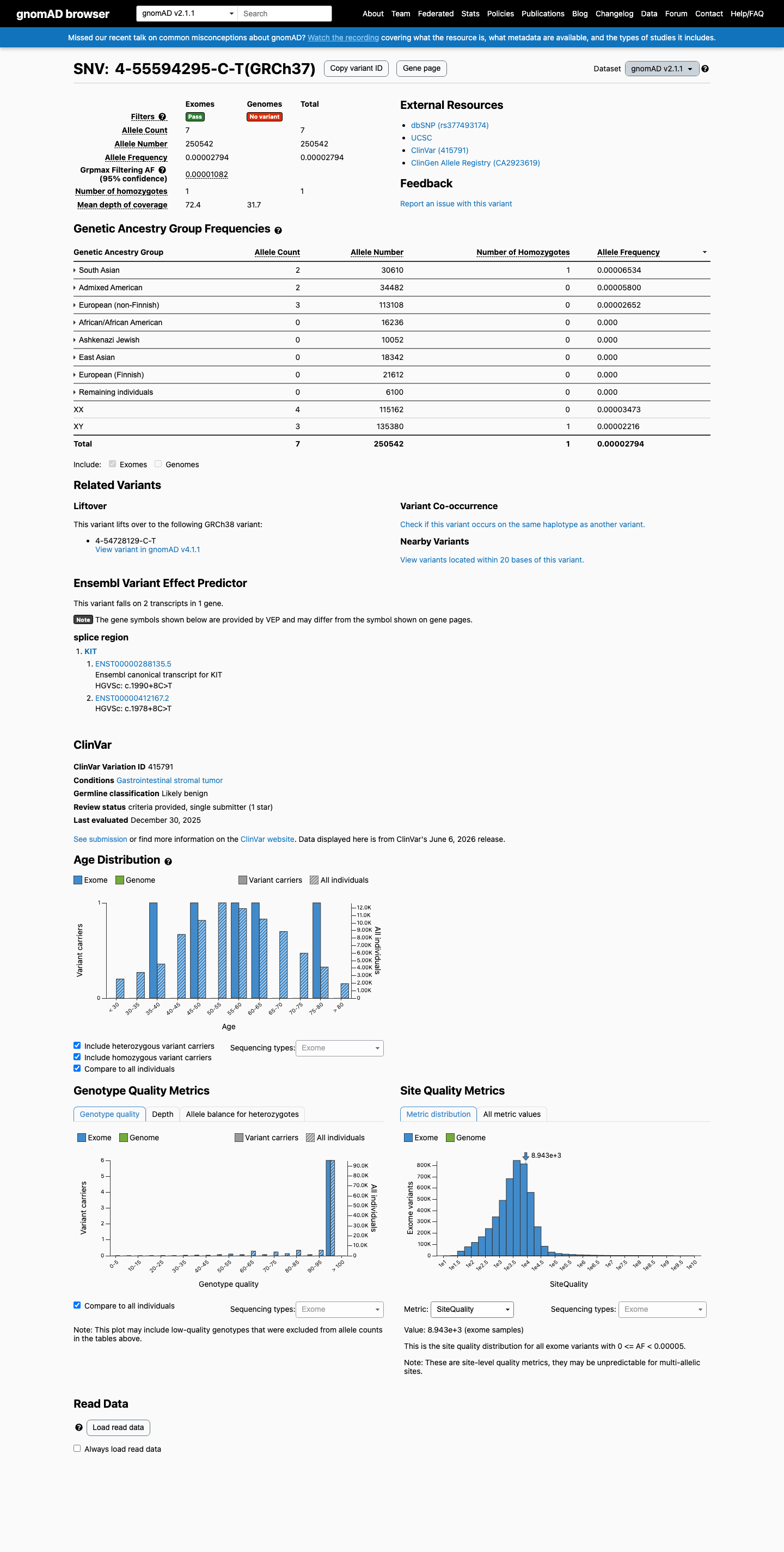
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.77833e-05; MAF= 0.00178%, 28/1574512 alleles, homozygotes = 1) and has highest observed frequency in the South Asian population (AF= 8.85936e-05; MAF= 0.00886%, 8/90300 alleles, homozygotes = 1); grpmax FAF= 4.401e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 2.79394e-05; MAF= 0.00279%, 7/250542 alleles, homozygotes = 1) and has highest observed frequency in the South Asian population (AF= 6.53381e-05; MAF= 0.00653%, 2/30610 alleles, homozygotes = 1); grpmax FAF= 1.082e-05.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0018%
· 28 / 1,574,512
1 hom · FAF 0.0044%
1 hom · FAF 0.0044%
South Asian 8 / 90,300 |
0.0089% 1 hom |
Admixed American 2 / 59,918 |
0.0033% |
European (non-Finnish) 18 / 1,144,066 |
0.0016% |
+ 7 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0028%
· 7 / 250,542
1 hom · FAF 0.0011%
1 hom · FAF 0.0011%
South Asian 2 / 30,610 |
0.0065% 1 hom |
Admixed American 2 / 34,482 |
0.0058% |
European (non-Finnish) 3 / 113,108 |
0.0027% |
+ 5 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
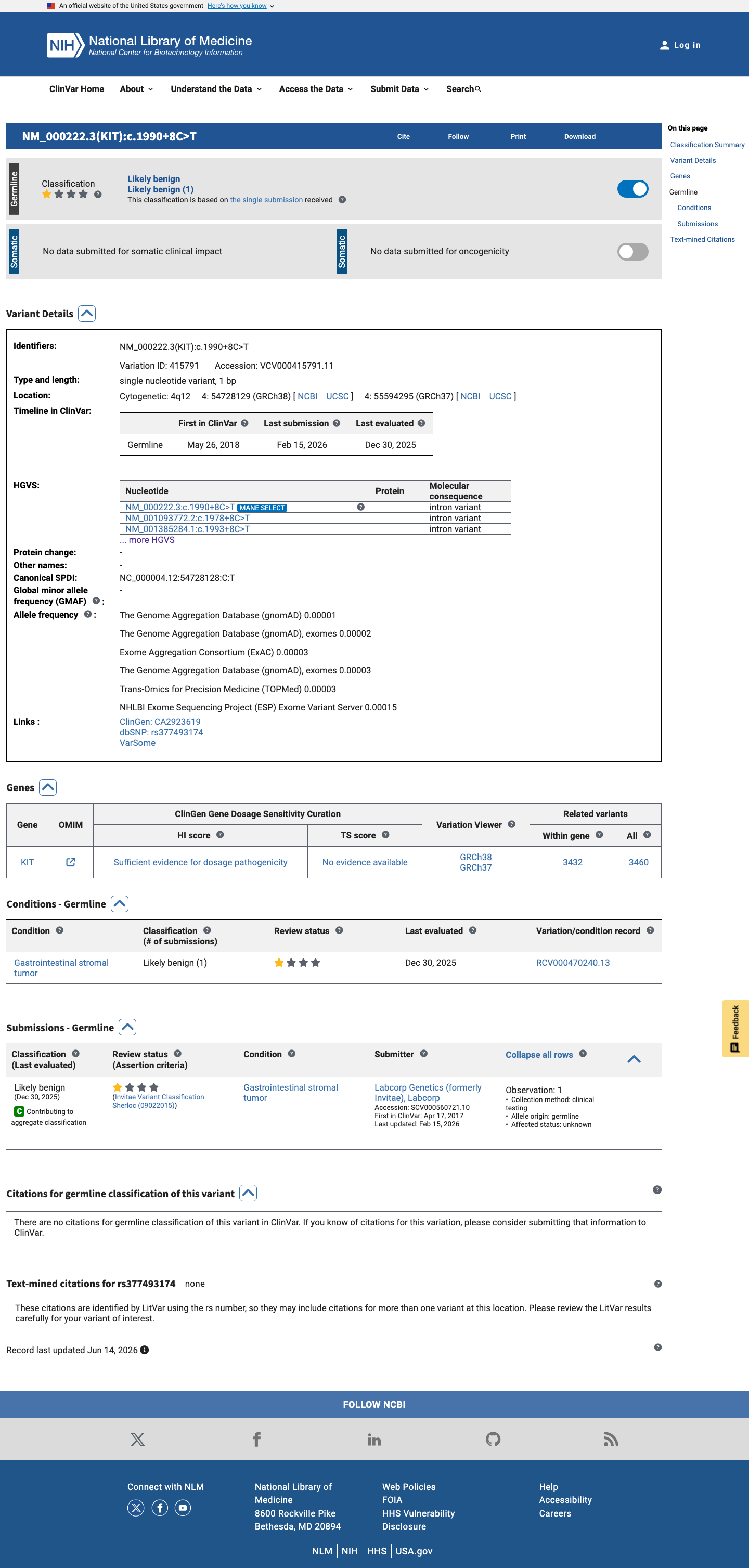
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory). (ClinVarID = 415791)
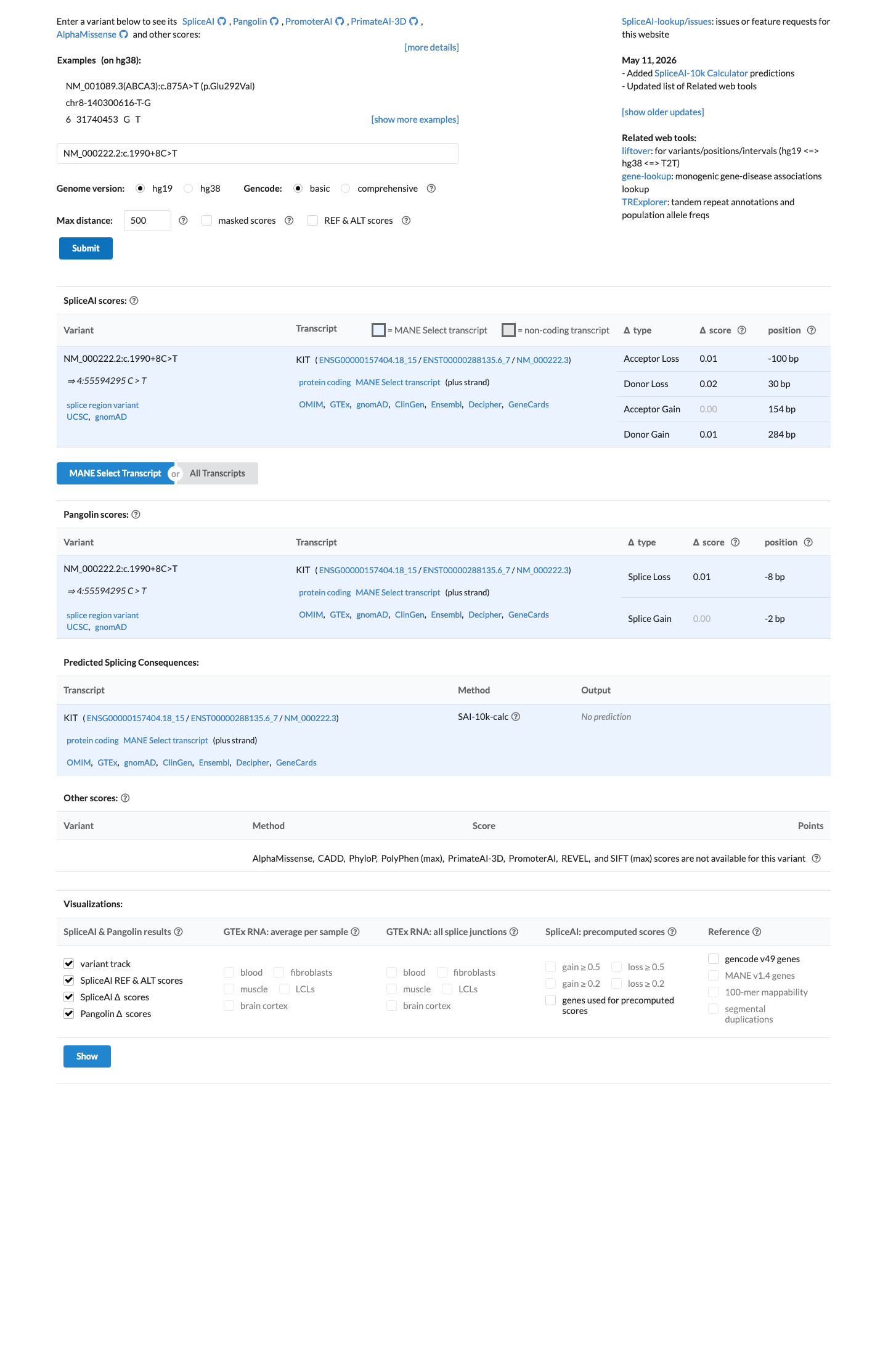
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
23852704 ↗
Tumor markers in colorectal cancer, gastric cancer and gastrointestinal stromal cancers: European group on tumor markers 2014 guidelines update.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
22685257 ↗
The UK NEQAS for Molecular Genetics scheme for gastrointestinal stromal tumour: findings and recommendations following four rounds of circulation.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR