NM_000222.3:c.-14T>A is a 5'UTR substitution in the KIT gene, located 14 nucleotides upstream of the ATG start codon. This variant is present at appreciable frequency in population databases: gnomAD v2.1 allele frequency 0.35% (977/281,098 alleles, 2 homozygotes) and gnomAD v4.1 allele frequency 0.53% (8,496/1,613,094 alleles, 35 homozygotes), with the highest subpopulation frequency of 0.65% in the European (non-Finnish) population (BS1).1 The observation of 35 homozygous individuals in gnomAD v4.1 is incompatible with a highly penetrant dominant germline disorder such as familial GIST or piebaldism (BS2).2 SpliceAI predicts no splicing impact (max delta score = 0.00); no computational evidence suggests a deleterious effect (BP4).3 ClinVar classifies this variant as Likely benign based on submissions from multiple clinical laboratories; 4 of 5 submitters concluded benign or likely benign (BP6).4 This variant has been reported in COSMIC (COSV55412424) in 2 somatic cancer samples, which is consistent with a benign germline variant occasionally detected in tumor sequencing. No variant-specific functional studies, de novo occurrences, segregation data, or pathogenic assertions from reputable sources were identified for this variant.
KIT
Final classification
Benign
KIT c.-14T>A · p.?
KIT
NM_000222.3:c.-14T>A is a 5'UTR substitution in the KIT gene, located 14 nucleotides upstream of the ATG start codon.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong, BS2 strong, BP4 supporting, BP6 supporting; combination = 2 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BS1BS2BP4BP6
Benign
KIT c.-14T>A
BS1 + BS2 + BP4 + BP6
→
Benign
Gene diagram
· NM_000222.3 · variants mapped to exon structure
KIT
NM_000222.3
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 15 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
BS1 is met at strong level: allele frequency exceeds the 0.3% threshold in the European (non-Finnish) population. gnomAD v2.1 NFE AF = 0.592% and gnomAD v4.1 NFE AF = 0.654%, both substantially above the expected frequency for rare KIT-related germline disorders (familial GIST, piebaldism).
gnomAD v2.1: NFE AF = 0.592% (758/128014 alleles1 homozygote)
✓
BS2
strong
Benign
BS2 is met at strong level: 35 homozygotes are observed in gnomAD v4.1 and 2 homozygotes in gnomAD v2.1. The presence of homozygous individuals in a general population database is inconsistent with a highly penetrant dominant germline disorder such as familial GIST or piebaldism, where homozygosity would be expected to be lethal or cause severe early-onset disease.
gnomAD v2.1: 2 homozygotes across 281098 alleles. gnomAD v4.1: 35 homozygotes across 1613
✓
BP4
supporting
Benign
BP4 is met at supporting level: SpliceAI predicts no splicing impact (max delta score = 0.00) for this 5'UTR variant. No other computational predictors suggest a deleterious effect. Multiple lines of computational evidence support a benign interpretation.
SpliceAI max delta = 0.00all splice site gain/loss scores null. No REVELBayesDel
✓
BP6
supporting
Benign
BP6 is met at supporting level: ClinVar reports a consensus classification of Likely benign from multiple clinical laboratories (3 Likely benign, 1 Benign, 1 Uncertain significance). Multiple independent submitters using clinical testing methodology have reached a benign or likely benign conclusion.
ClinVar Variation ID 255567: 5 total submissions — Likely benign (3 clinical labs: PreventionGeneticsIllumina ×2)Benign (1 clinical lab: Illumina)
Assessed · not applied
Pathogenic
PS2
No de novo occurrence data are available for this variant; PS2 requires confirmed de novo status with both maternity and paternity confirmed.
PS3
No well-established in vitro or in vivo functional studies have been identified for this 5'UTR variant; PS3 requires variant-specific functional evidence demonstrating a damaging effect on the gene or gene product.
PS4
The variant is present at 0.35–0.53% allele frequency in gnomAD population controls, inconsistent with a rare pathogenic variant; prevalence in affected individuals does not exceed that in the general population.
PM1
This variant is a 5'UTR substitution located upstream of the coding sequence; it does not reside in a known mutational hotspot or critical functional domain of KIT.
PM2
PM2 requires absence or very low frequency in population databases (<0.1%).
PM6
No de novo observation has been reported for this variant; PM6 requires a de novo occurrence with maternity and paternity not confirmed.
PP1
No co-segregation data are available for this variant; PP1 requires co-segregation with disease in multiple affected family members.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No patient phenotype or family history data are available; PP4 requires that the patient's phenotype or family history is highly specific for a disease with a single genetic etiology.
PP5
Reputable sources (ClinVar) do not report this variant as pathogenic; the ClinVar consensus classification is Likely benign (3 laboratories) with additional submissions of Benign (1) and Uncertain significance (1).
Benign
BA1
BA1 requires allele frequency >1% in any population.
BS3
No well-established in vitro or in vivo functional studies demonstrating no damaging effect on protein function or splicing have been identified for this 5'UTR variant.
BS4
No co-segregation data are available; BS4 requires lack of segregation with disease in affected family members.
BP2
No evidence that this variant has been observed in trans with a pathogenic variant or in cis with a pathogenic variant in any individual.
BP5
No evidence that this variant has been observed in a case with an alternate molecular basis for disease; BP5 requires identification of a different confirmed pathogenic variant explaining the phenotype in the same patient.
N/A · 6
PVS1 · PS1 · PM5 · PP2 · BP1 · BP7
Research & evidence
Population frequency · supports benign
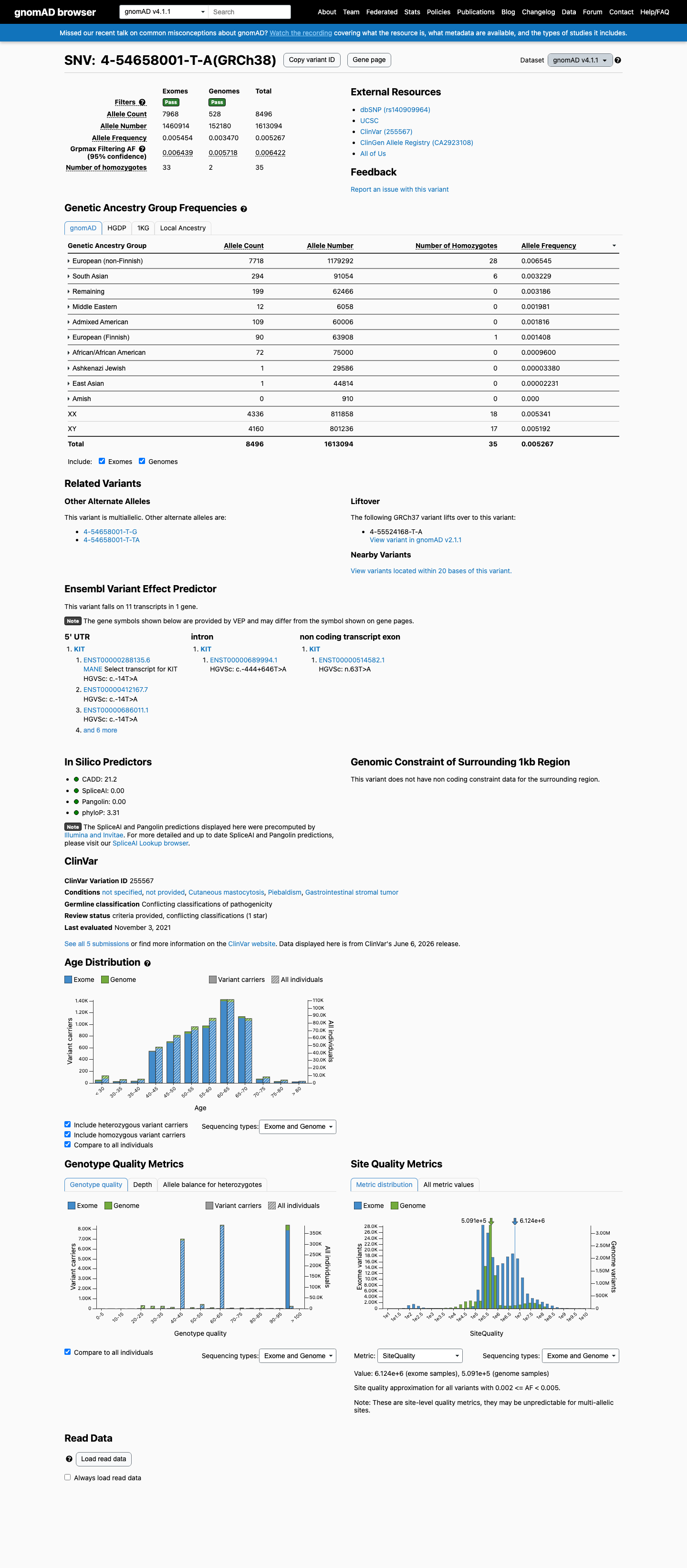
gnomAD v4.1
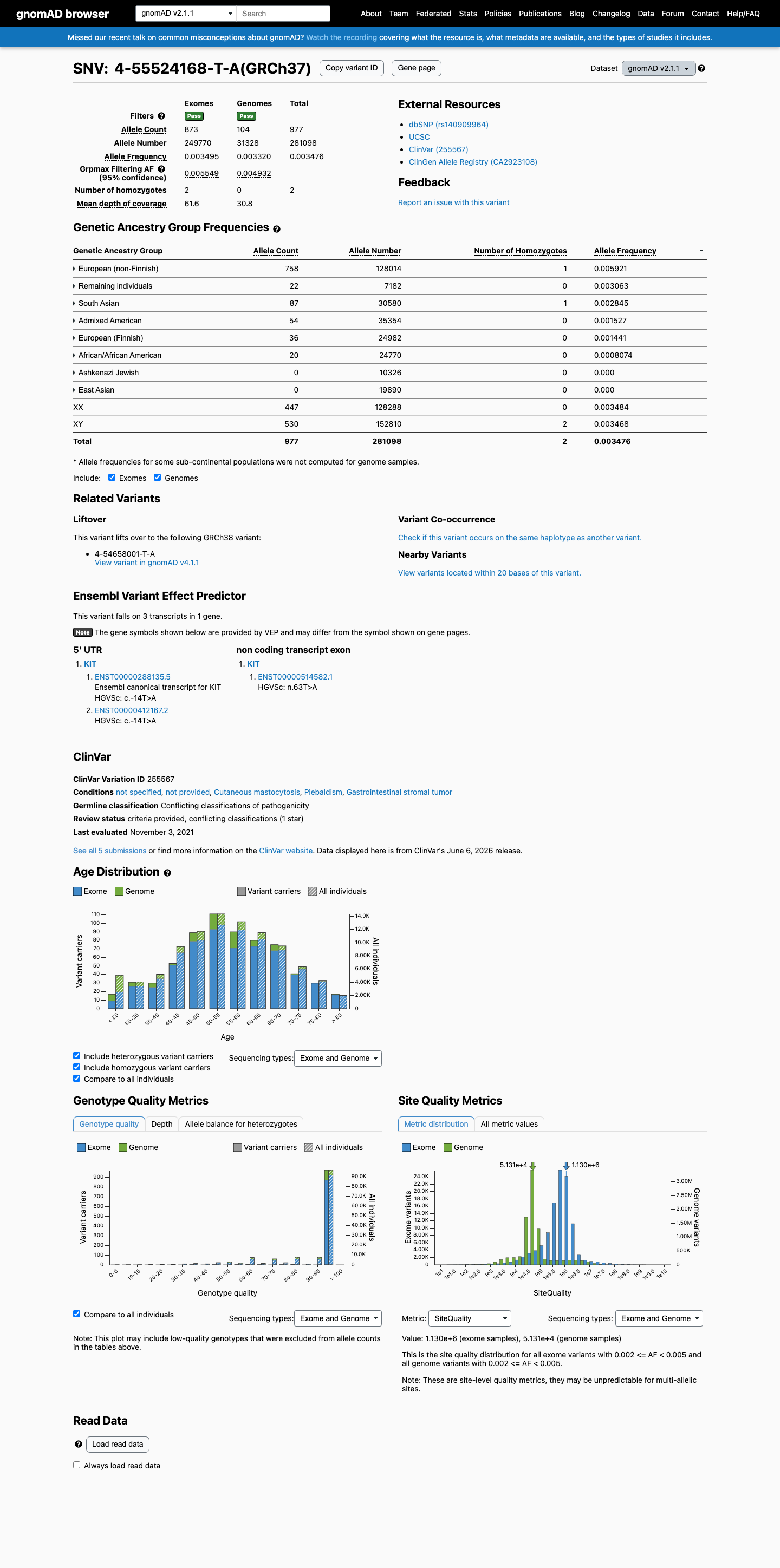
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.0052669; MAF= 0.52669%, 8496/1613094 alleles, homozygotes = 35) and has highest observed frequency in the European (non-Finnish) population (AF= 0.0065446; MAF= 0.65446%, 7718/1179292 alleles, homozygotes = 28); grpmax FAF= 0.00642186.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00347566; MAF= 0.34757%, 977/281098 alleles, homozygotes = 2) and has highest observed frequency in the European (non-Finnish) population (AF= 0.00592123; MAF= 0.59212%, 758/128014 alleles, homozygotes = 1); grpmax FAF= 0.00554939.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0056595559425337396, 104/18376 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.53%
· 8496 / 1,613,094
35 hom · FAF 0.64%
35 hom · FAF 0.64%
European (non-Finnish) 7718 / 1,179,292 |
0.65% 28 hom |
South Asian 294 / 91,054 |
0.32% 6 hom |
Remaining individuals 199 / 62,466 |
0.32% |
Middle Eastern 12 / 6,058 |
0.2% |
Admixed American 109 / 60,006 |
0.18% |
European (Finnish) 90 / 63,908 |
0.14% 1 hom |
African/African American 72 / 75,000 |
0.096% |
Ashkenazi Jewish 1 / 29,586 |
0.0034% |
East Asian 1 / 44,814 |
0.0022% |
+ 1 not observed (Amish)
gnomAD v2.1
0.35%
· 977 / 281,098
2 hom · FAF 0.55%
2 hom · FAF 0.55%
European (non-Finnish) 758 / 128,014 |
0.59% 1 hom |
Remaining individuals 22 / 7,182 |
0.31% |
South Asian 87 / 30,580 |
0.28% 1 hom |
Admixed American 54 / 35,354 |
0.15% |
European (Finnish) 36 / 24,982 |
0.14% |
African/African American 20 / 24,770 |
0.081% |
+ 2 not observed (Ashkenazi Jewish, East Asian)
gnomAD Canada 🇨🇦
0.57%
· 104 / 18,376
0 hom · FAF 0.62%
0 hom · FAF 0.62%
Middle Eastern 4 / 142 |
2.8% |
European (non-Finnish) 87 / 11,706 |
0.74% |
Remaining individuals 8 / 1,138 |
0.7% |
South Asian 4 / 1,358 |
0.29% |
African/African American 1 / 1,016 |
0.098% |
+ 4 not observed (Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish))
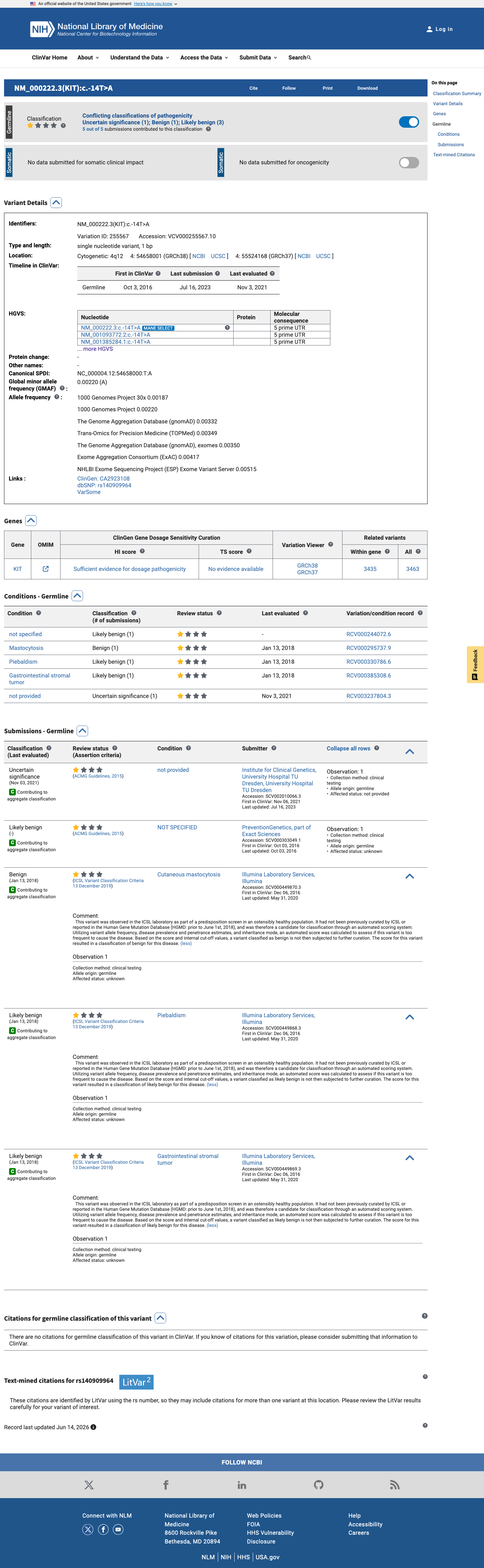
ClinVar
This variant has been reported in ClinVar as Likely benign (3 clinical laboratories) and as Uncertain significance (1 clinical laboratory) and as Benign (1 clinical laboratory). (ClinVarID = 255567)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
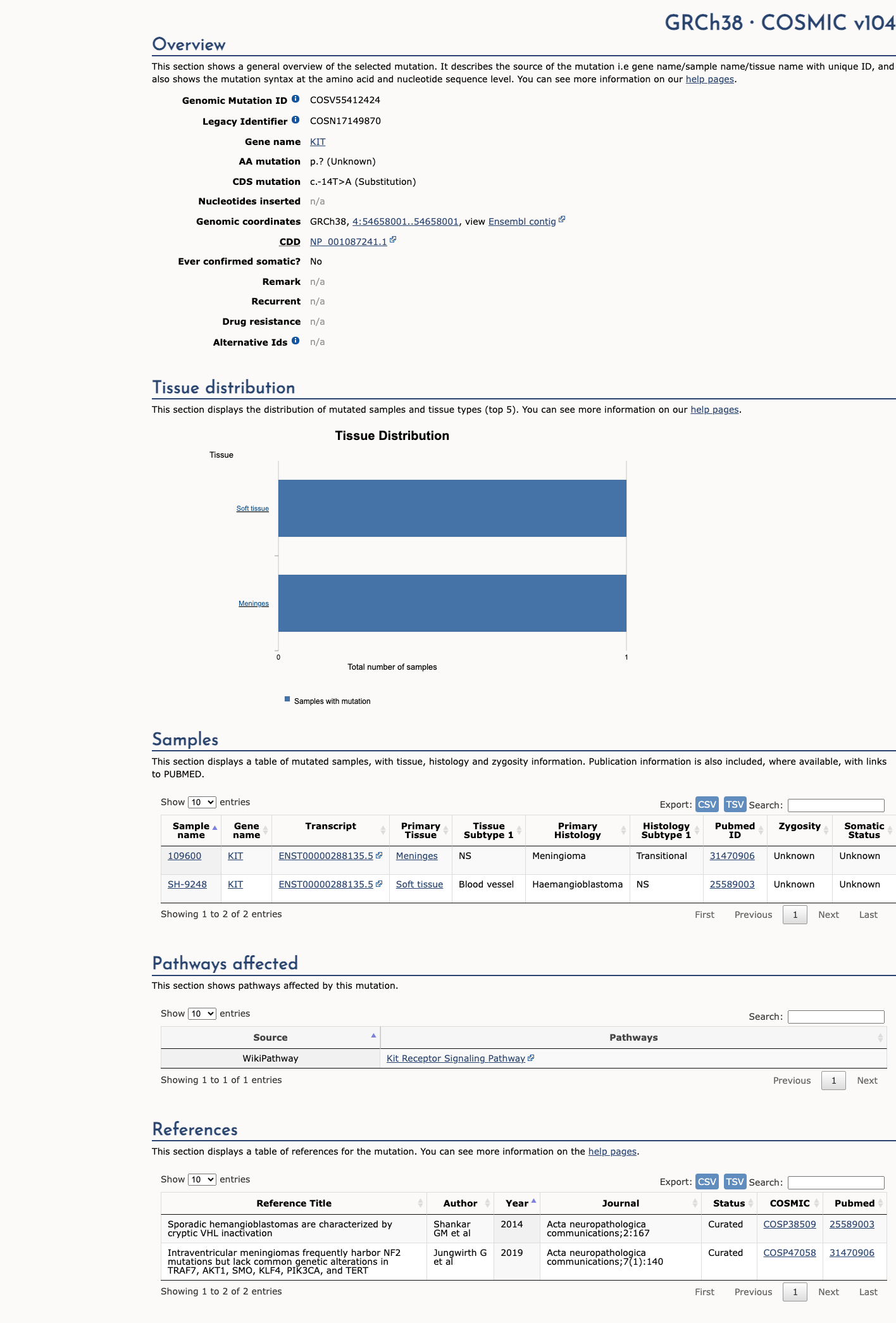
COSMIC
Somatic evidence
COSMIC
This variant has previously been reported in somatic cancers (COSMIC; COSV55412424, n = 2 times).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
23852704 ↗
Tumor markers in colorectal cancer, gastric cancer and gastrointestinal stromal cancers: European group on tumor markers 2014 guidelines update.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
22685257 ↗
The UK NEQAS for Molecular Genetics scheme for gastrointestinal stromal tumour: findings and recommendations following four rounds of circulation.
CLINVAR