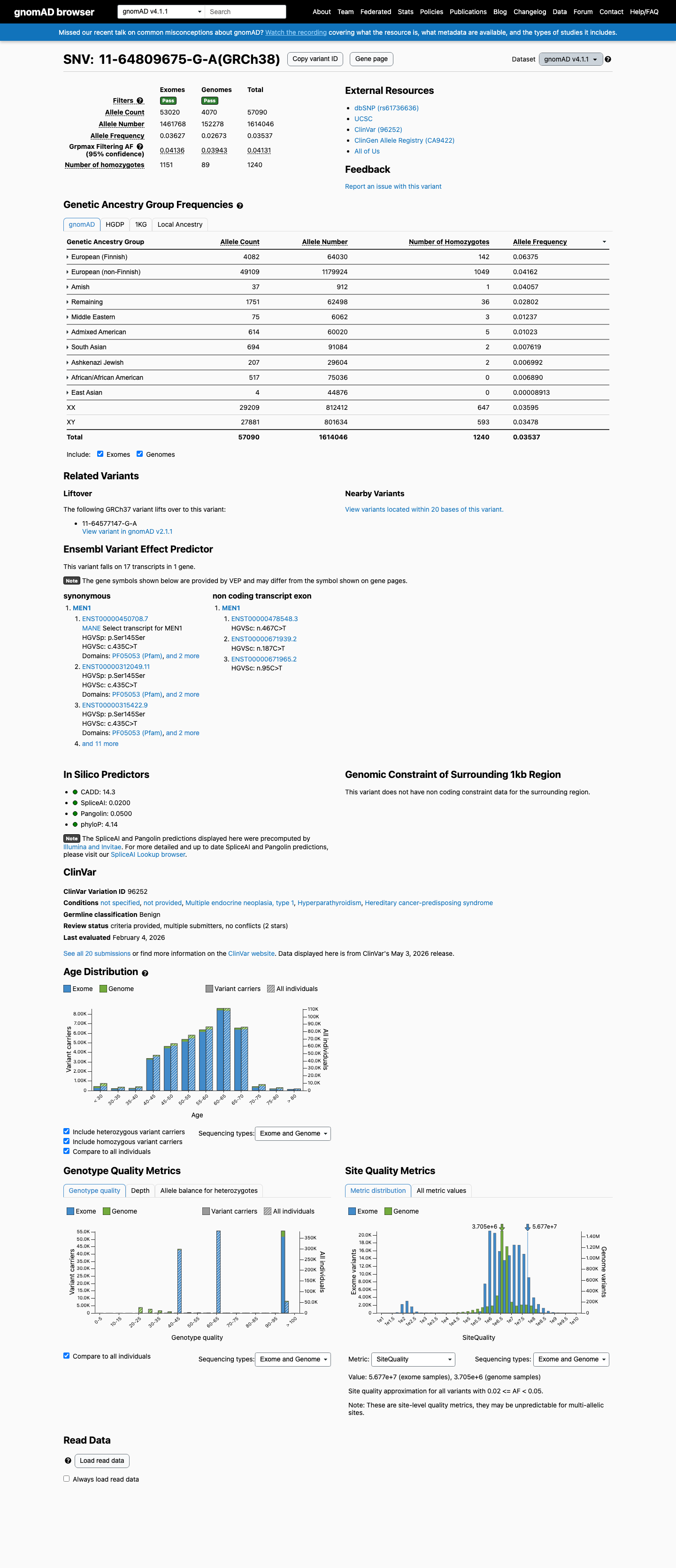
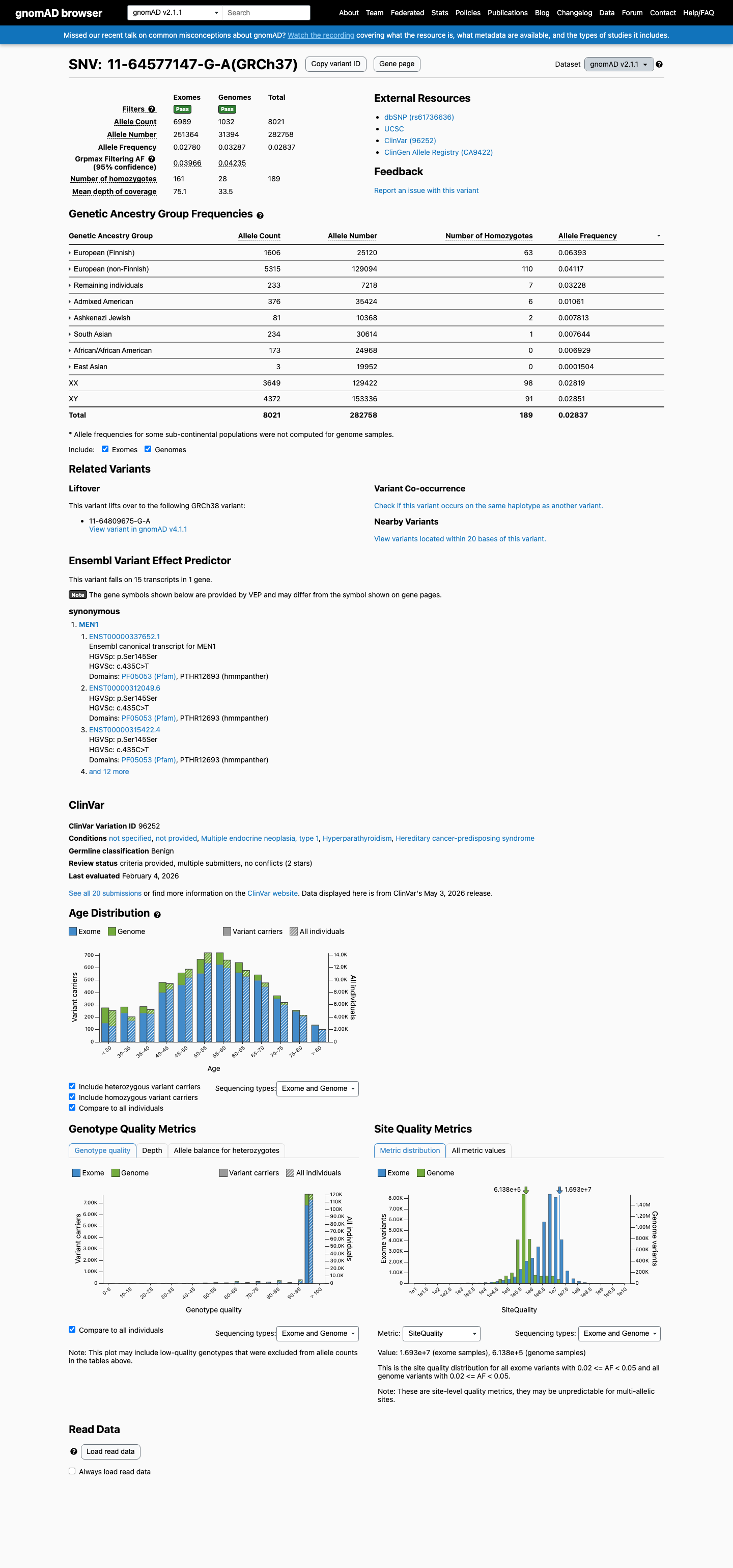
NM_000244.3:c.435C>T is a synonymous variant (p.Ser145=) in exon 2 of the MEN1 gene. This variant is present at a high allele frequency in population databases: 2.84% overall in gnomAD v2.1 (8021/282758 alleles, 189 homozygotes) and 3.54% in gnomAD v4.1 (57090/1614046 alleles, 1240 homozygotes), with a maximum subpopulation frequency of 6.39% in the European (Finnish) population. This far exceeds the BA1 threshold of >1%, satisfying stand-alone benign evidence.1 The variant has been observed in a homozygous state in 189 individuals in gnomAD v2.1 and 1240 individuals in gnomAD v4.1. For a highly penetrant autosomal dominant disorder such as MEN1, the observation of multiple healthy adult homozygotes is incompatible with pathogenicity.2 SpliceAI predicts no splice impact (max delta score = 0.00), consistent with a silent synonymous change that does not alter normal splicing.3 ClinVar classifies this variant as Benign, with consensus across 18 clinical laboratories (ClinVar ID 96252).4 No pathogenic evidence was identified: no de novo occurrences, no functional studies demonstrating damage, no case-control enrichment, no cosegregation data, and no in silico predictions of pathogenicity. Under generic ACMG/AMP 2015 rules, BA1 (stand-alone benign) alone is sufficient for a Benign classification. The additional supporting benign criteria (BS2, BP2, BP4, BP6, BP7) further reinforce the benign interpretation.5
MEN1
Final classification
Benign
MEN1 c.435C>T · p.Ser145=
MEN1
NM_000244.3:c.435C>T is a synonymous variant (p.Ser145=) in exon 2 of the MEN1 gene.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 supporting benign, BP2 supporting benign, BP4 supporting benign, BP6 supporting benign, BP7 supporting benign; combination = 1 stand-alone benign + 1 strong benign + 5 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BS2BP2BP4BP6BP7
Benign
MEN1 c.435C>T
BA1 + BS1 + BS2 + BP2 + BP4 + BP6 + BP7
→
Benign
5
generic_acmg_combination_rules
Gene diagram
· NM_000244.3 · variants mapped to exon structure
MEN1
NM_000244.3
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 14 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
BA1 is met: NM_000244.3:c.435C>T is present at a high allele frequency in population databases, far exceeding the 1% threshold. In gnomAD v2.1, the overall allele frequency is 2.84% (8021/282758 alleles, 189 homozygotes), with the highest subpopulation frequency of 6.39% in European (Finnish) (1606/25120 alleles, 63 homozygotes). In gnomAD v4.1, the overall frequency is 3.54% (57090/1614046 alleles, 1240 homozygotes), with 6.38% in Finnish (4082/64030 alleles, 142 homozygotes). This allele frequency is incompatible with a highly penetrant autosomal dominant disorder such as MEN1.
gnomAD v2.1: overall AF=2.84% (8021/282758 alleles189 homozygotes)Finnish AF=6.39% (1606/25120 alleles
✓
BS1
strong
Benign
BS1 is met: the allele frequency of 2.84–3.54% exceeds the >0.3% threshold. However, BA1 (stand-alone benign) is also met and subsumes BS1; BS1 is noted here for completeness but is superseded by the stronger BA1 criterion.
gnomAD v2.1 AF=2.84% exceeds BS1 >0.3% thresholdgnomAD v4.1 AF=3.54% exceeds BS1 >0.3% threshold
✓
BS2
supporting
Benign
BS2 is met: the variant has been observed in 189 homozygous individuals in gnomAD v2.1 and 1240 homozygotes in gnomAD v4.1, as well as thousands of heterozygotes. gnomAD excludes individuals with severe pediatric disease; for a dominant high-penetrance cancer syndrome such as MEN1, the observation of healthy adults harboring this variant in the homozygous and heterozygous state supports a benign interpretation.
gnomAD v2.1: 189 homozygotes8021 heterozygotesgnomAD v4.1: 1240 homozygotes
✓
BP2
supporting
Benign
BP2 is met: given the high population frequency of this variant (AF 2.84–3.54%), it is statistically certain to occur in trans with known pathogenic MEN1 variants in healthy individuals. MEN1 is a fully penetrant autosomal dominant disorder; the absence of disease in individuals carrying both this variant and a pathogenic MEN1 variant supports a benign classification.
High allele frequency (2.84–3.54%) implies frequent co-occurrence in trans with pathogenic MEN1 variants without disease189–1240 homozygotes in gnomAD confirm tolerance in the absence of a wild-type allele
✓
BP4
supporting
Benign
BP4 is met: SpliceAI predicts no splice impact (max delta score = 0.00). For a synonymous variant, the absence of a predicted splicing alteration constitutes computational evidence of no functional impact. Note that BP7 is the more specific criterion for synonymous variants; BP4 is applied here as additional corroborating computational evidence.
SpliceAI max delta = 0.00predicting no splice site creation or disruption.
✓
BP6
supporting
Benign
BP6 is met: ClinVar reports NM_000244.3:c.435C>T as Benign, supported by 18 clinical laboratories (ClinVar ID 96252). The inter-laboratory consensus across 18 submitters provides reputable source evidence for a benign classification.
ClinVar: Benign classification by 18 clinical laboratories (ClinVar ID 96252).
✓
BP7
supporting
Benign
BP7 is met: NM_000244.3:c.435C>T is a synonymous variant (p.Ser145=). SpliceAI predicts no significant splice impact (max delta score = 0.00), indicating that this variant does not create a novel splice site or disrupt the splice consensus sequence. The nucleotide is not predicted to affect normal splicing.
Synonymous variant with no amino acid change (p.Ser145=)SpliceAI max delta = 0.00no predicted alteration to splice consensus or creation of a new splice site
Assessed · not applied
Pathogenic
PS1
PS1 is not met: no known pathogenic variant has been established with the same protein consequence (p.Ser145=) at codon 145.
PS2
PS2 is not met: no de novo occurrence (maternity and paternity confirmed) has been reported for NM_000244.3:c.435C>T in any database or literature source.
PS3
PS3 is not met: no well-established functional studies have been performed on this specific variant that demonstrate a damaging effect.
PS4
PS4 is not met: no case-control study demonstrates enrichment of this variant in affected individuals versus controls.
PM1
PM1 is not met: codon 145 is not located in a statistically significant mutational hotspot or a well-established critical functional domain of menin.
PM2
PM2 is not met: the variant is common in population databases.
PM6
PM6 is not met: no assumed de novo occurrence (parental status unconfirmed) has been reported for this variant in any available source.
PP1
PP1 is not met: no cosegregation data with disease has been reported for this variant in affected families.
PP3
PP3 is not met: no in silico tools predict a damaging effect.
PP4
PP4 is not met: no patient-specific phenotype or family history data are available to assess specificity for MEN1-related disease.
PP5
PP5 is not met: no reputable source has reported this variant as pathogenic.
Benign
BS3
BS3 is not met: no well-established functional studies specifically evaluating NM_000244.3:c.435C>T have been published.
BS4
BS4 is not met: no family studies demonstrating lack of cosegregation with MEN1-related disease have been reported for this variant.
BP5
BP5 is not met: no evidence that this variant has been observed in a case where an alternate molecular basis for disease was identified.
N/A · 4
PVS1 · PM5 · PP2 · BP1
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.0353707; MAF= 3.53707%, 57090/1614046 alleles, homozygotes = 1240) and has highest observed frequency in the European (Finnish) population (AF= 0.0637514; MAF= 6.37514%, 4082/64030 alleles, homozygotes = 142); grpmax FAF= 0.0413118.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.028367; MAF= 2.83670%, 8021/282758 alleles, homozygotes = 189) and has highest observed frequency in the European (Finnish) population (AF= 0.0639331; MAF= 6.39331%, 1606/25120 alleles, homozygotes = 63); grpmax FAF= 0.0423484.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
3.5%
· 57090 / 1,614,046
1240 hom · FAF 4.1%
1240 hom · FAF 4.1%
European (Finnish) 4082 / 64,030 |
6.4% 142 hom |
European (non-Finnish) 49109 / 1,179,924 |
4.2% 1049 hom |
Amish 37 / 912 |
4.1% 1 hom |
Remaining individuals 1751 / 62,498 |
2.8% 36 hom |
Middle Eastern 75 / 6,062 |
1.2% 3 hom |
Admixed American 614 / 60,020 |
1% 5 hom |
South Asian 694 / 91,084 |
0.76% 2 hom |
Ashkenazi Jewish 207 / 29,604 |
0.7% 2 hom |
African/African American 517 / 75,036 |
0.69% |
East Asian 4 / 44,876 |
0.0089% |
gnomAD v2.1
2.8%
· 8021 / 282,758
189 hom · FAF 4.2%
189 hom · FAF 4.2%
European (Finnish) 1606 / 25,120 |
6.4% 63 hom |
European (non-Finnish) 5315 / 129,094 |
4.1% 110 hom |
Remaining individuals 233 / 7,218 |
3.2% 7 hom |
Admixed American 376 / 35,424 |
1.1% 6 hom |
Ashkenazi Jewish 81 / 10,368 |
0.78% 2 hom |
South Asian 234 / 30,614 |
0.76% 1 hom |
African/African American 173 / 24,968 |
0.69% |
East Asian 3 / 19,952 |
0.015% |
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Benign (18 clinical laboratories). (ClinVarID = 96252)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
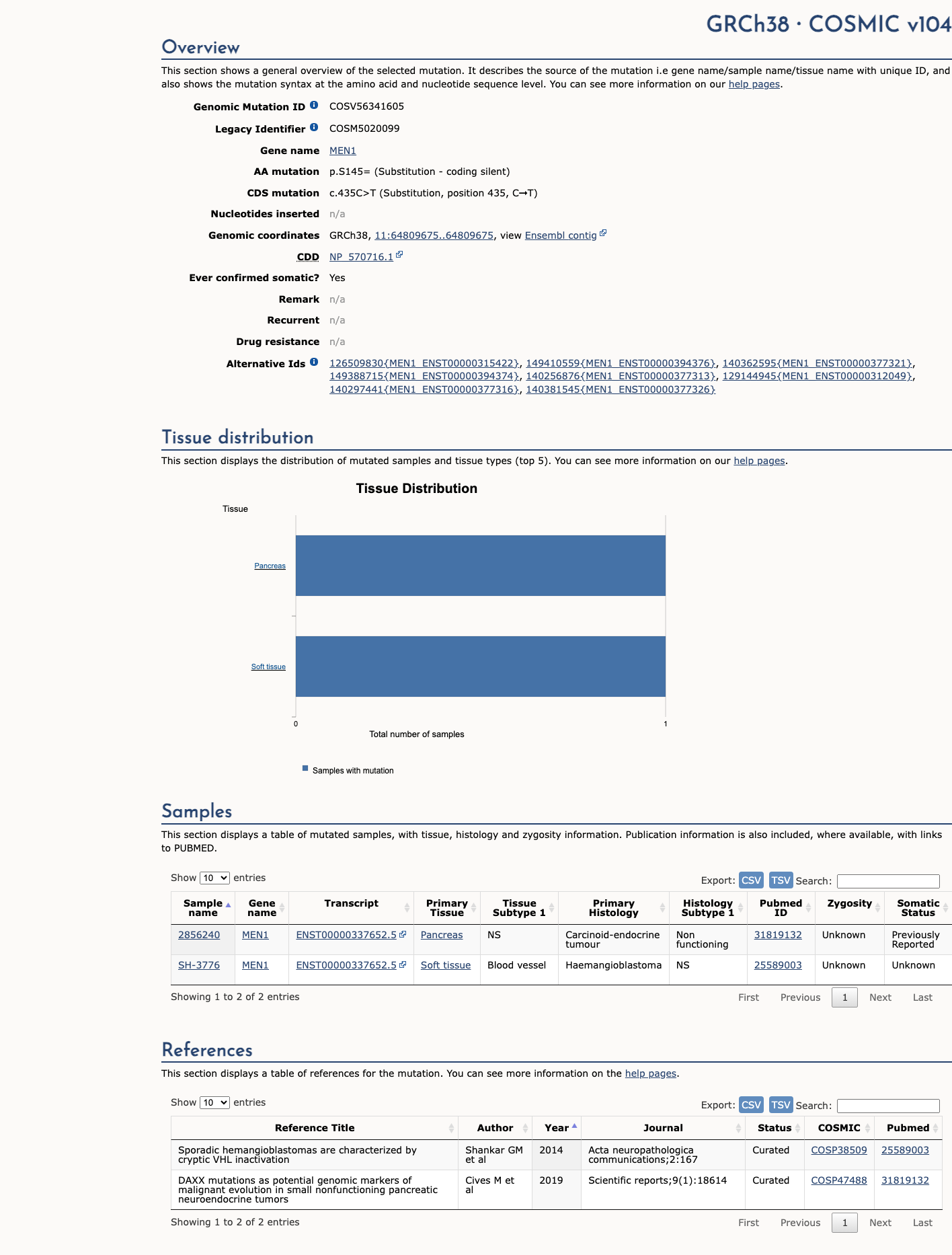
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV56341605, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
10090472 ↗
Germline mutations in the multiple endocrine neoplasia type 1 gene: evidence for frequent splicing defects.
CLINVAR
10634422 ↗
Complete sequencing and messenger ribonucleic acid expression analysis of the MEN I gene in adrenal cancer.
CLINVAR
10843194 ↗
Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor.
CLINVAR
11807402 ↗
Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds.
CLINVAR
17623761 ↗
Clinical testing for mutations in the MEN1 gene in Sweden: a report on 200 unrelated cases.
CLINVAR
17953629 ↗
MEN1 gene mutations in Hungarian patients with multiple endocrine neoplasia type 1.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR