Classification rationale
BA1BP4
Benign
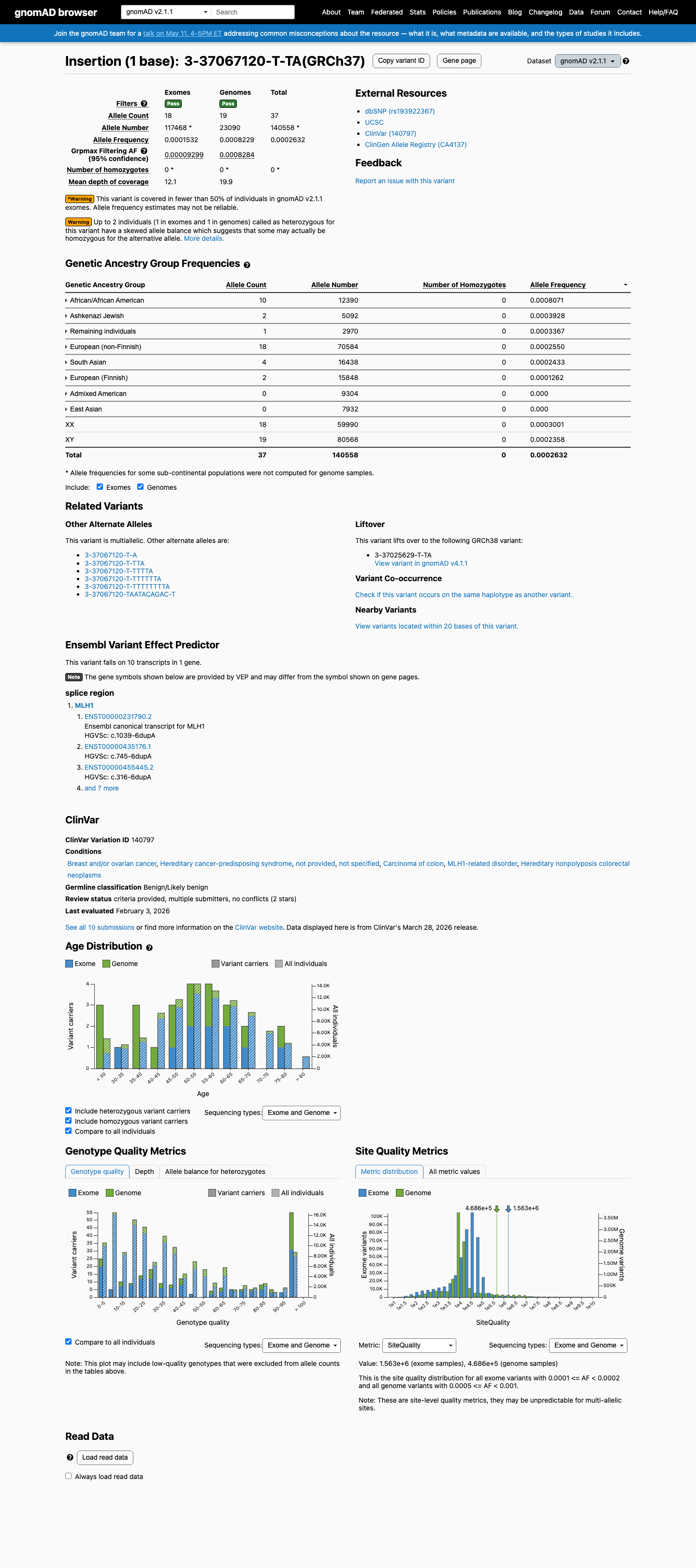
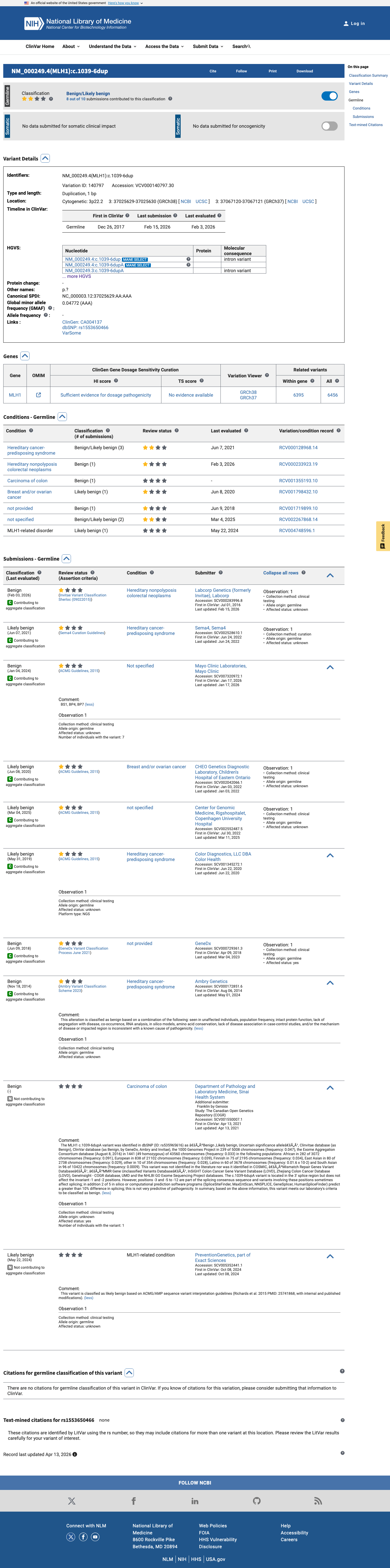
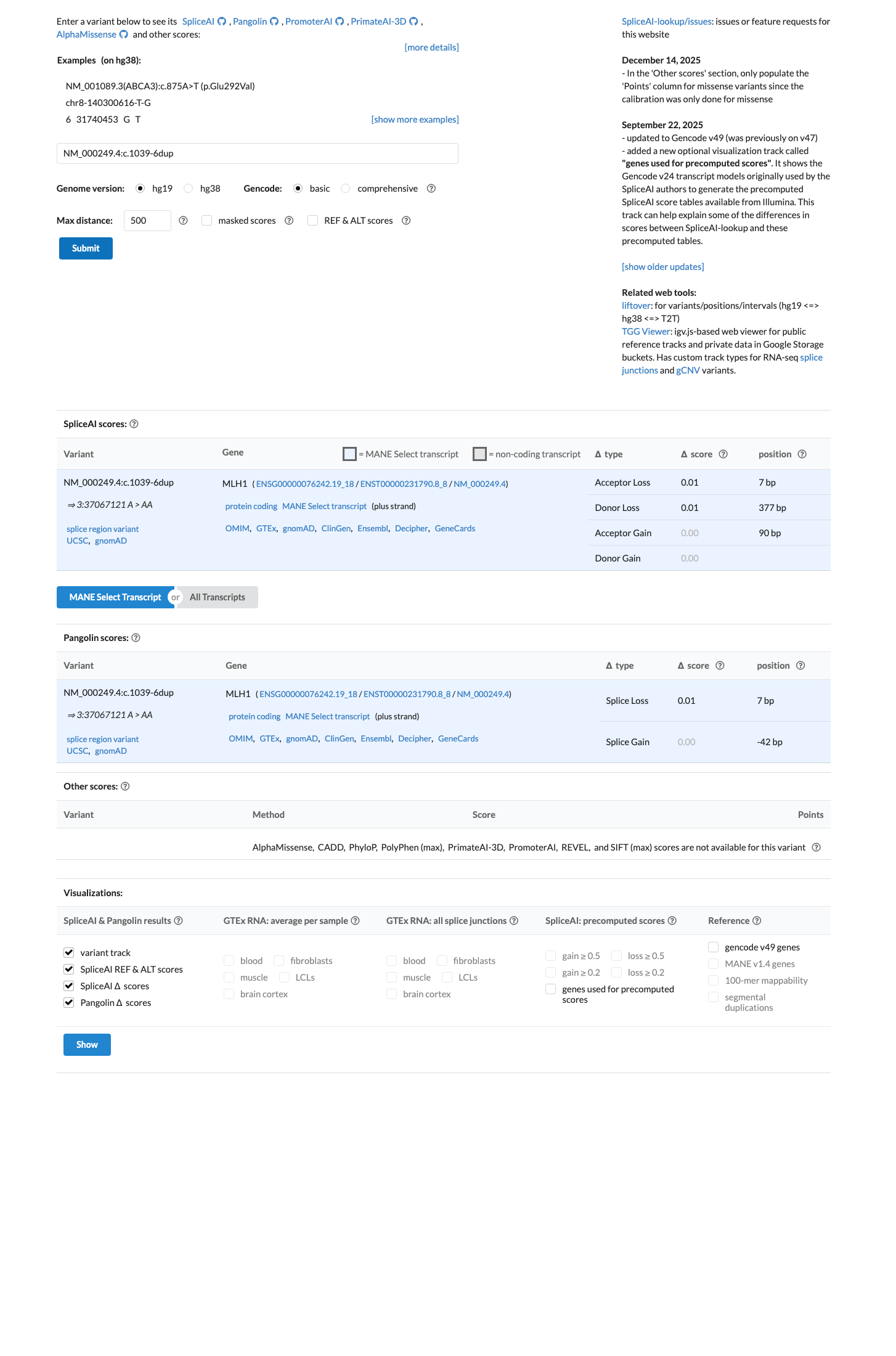
MLH1 c.1039-6dup
The MLH1 c.1039-6dup (p.?) variant has been reported in ClinVar as benign by 5 clinical laboratories and likely benign by 4 clinical laboratories.1 This variant is present in population databases; in gnomAD v4.1 the overall allele frequency is 0.000530357 and the grpmax filtering allele frequency is 0.00117106, which is above the MLH1 VCEP BA1 threshold of 0.001.2 SpliceAI predicts no significant splice effect, with a maximum delta score of 0.01, which is below the BP4 no-impact threshold of 0.1 and below the PP3 splice-defect threshold of 0.2 in the MLH1 VCEP rules.3
BA1 + BP4
→
Benign