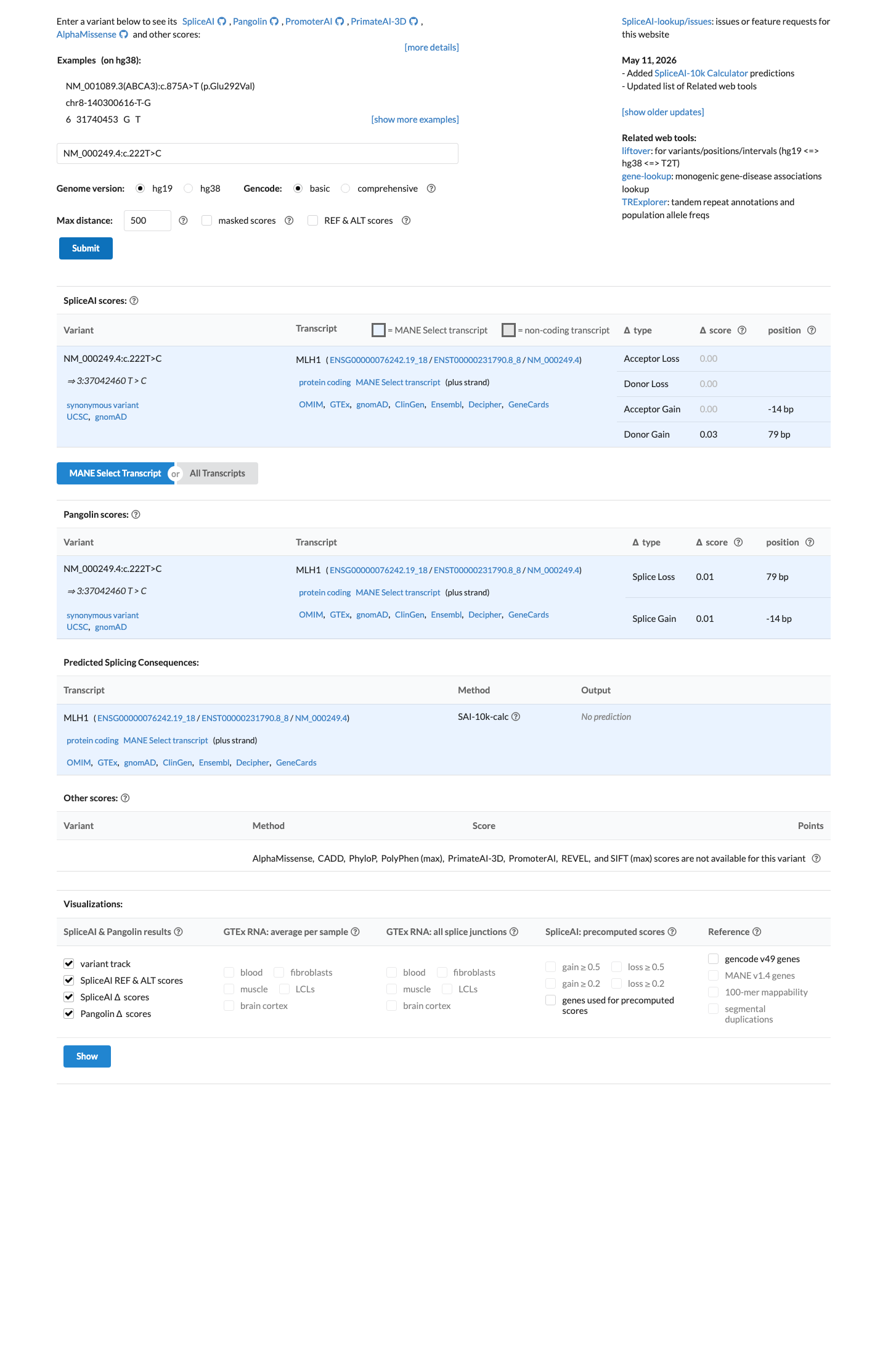
NM_000249.4:c.222T>C is a synonymous variant (p.Asp74=) in exon 3 of MLH1. SpliceAI predicts no splicing impact (max delta score 0.03), satisfying BP4_Supporting under InSiGHT MLH1 VCEP v2.0.0 rules for synonymous variants.1 The variant is located within the -21 exonic splice region (c.222 in exon 3, acceptor c.208), satisfying BP7_Supporting.2 An RNA splicing assay performed by a clinical diagnostic laboratory indicates this variant does not significantly alter splicing, consistent with a benign functional effect (BS3_Supporting).3 The variant is present in gnomAD v4.1 at a low frequency (37/1,595,534 alleles; AF=2.32e-05) that exceeds the VCEP PM2_Supporting threshold of <0.00002, therefore PM2 is not met. The frequency does not reach BS1 or BA1 thresholds.4 The variant has been reported in ClinVar as Likely benign by six clinical laboratories (ClinVar ID 237333).5 No evidence of pathogenicity was identified: PVS1, PS1, and PM5 are not applicable (synonymous); PS2/PM6, PP1, PP4, BS2, BS4, and BP5 lack data; PS3, PP3, BA1, and BS1 are not met.6
MLH1
Final classification
Likely Benign
MLH1 c.222T>C · p.Asp74=
MLH1
NM_000249.4:c.222T>C is a synonymous variant (p.Asp74=) in exon 3 of MLH1.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MLH1 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule19 (Benign.Supporting >=2) with applied criteria: BS3 supporting, BP4 supporting, BP7 supporting; maps to Likely Benign.
Classification rationale
BS3BP4BP7
Likely Benign
MLH1 c.222T>C
BS3 + BP4 + BP7
→
Likely Benign
Gene diagram
· NM_000249.4 · variants mapped to exon structure
MLH1
NM_000249.4
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 11 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS3
supporting
review
Benign
BS3_Supporting is met. An RNA splicing assay performed by a clinical diagnostic laboratory (Invitae, reported via ClinVar) indicates that NM_000249.4:c.222T>C does not significantly alter splicing. This is direct functional evidence of no splicing aberration for a synonymous variant where aberrant splicing would be the only plausible pathogenic mechanism. SpliceAI independently predicts no splicing impact (max delta 0.03). Under the VCEP BS3_Supporting rule, variant-specific proficient function in mRNA-based lab assays qualifies.
Invitae RNA splicing assay (ClinVar): no significant alteration of splicingSpliceAI max delta=0.03 (no predicted splice impact).
✓
BP4
supporting
Benign
BP4_Supporting is met. Under the VCEP rule for synonymous variants, BP4_Supporting applies when SpliceAI predicts no splicing impact with a delta score <=0.1. SpliceAI max delta for NM_000249.4:c.222T>C is 0.03, meeting this threshold. Additionally, REVEL score is 0.035 (strongly benign-leaning).
SpliceAI max delta=0.03 (<=0.1no predicted splice impact)REVEL=0.035.
✓
BP7
supporting
Benign
BP7_Supporting is met. NM_000249.4:c.222T>C is a synonymous variant located at nucleotide position 15 within exon 3 (c.208-c.306). This falls within the -21 position from the acceptor splice site (positions c.208-c.228), satisfying the VCEP BP7 rule for synonymous variants at or beyond -21/+7 (5'/3' exonic). SpliceAI also predicts no splicing impact (delta 0.03). BP7 and BP4 may both be applied.
Synonymous variant at c.222within -21 from exon 3 acceptor (c.208)SpliceAI delta=0.03.
Assessed · not applied
Pathogenic
PS2
No de novo occurrences of NM_000249.4:c.222T>C were identified in any database or publication.
PS3
PS3 is not met.
PM2
PM2_Supporting is not met.
PP1
No co-segregation data are available for this variant.
PP3
PP3 is not met.
PP4
No tumor MSI/IHC data specific to this variant are available.
Benign
BA1
BA1 is not met.
BS1
BS1 is not met.
BS2
No data on co-occurrence in trans with a known pathogenic MLH1 variant are available.
BS4
No segregation or lack-of-segregation data are available for this variant.
BP5
No tumor phenotype data (MSS status, MMR protein expression, BRAF/MLH1 methylation) are available for this variant.
N/A · 11
PVS1 · PS1 · PS4 · PM1 · PM5 · PM6 · PP2 · PP5 · BP1 · BP2 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
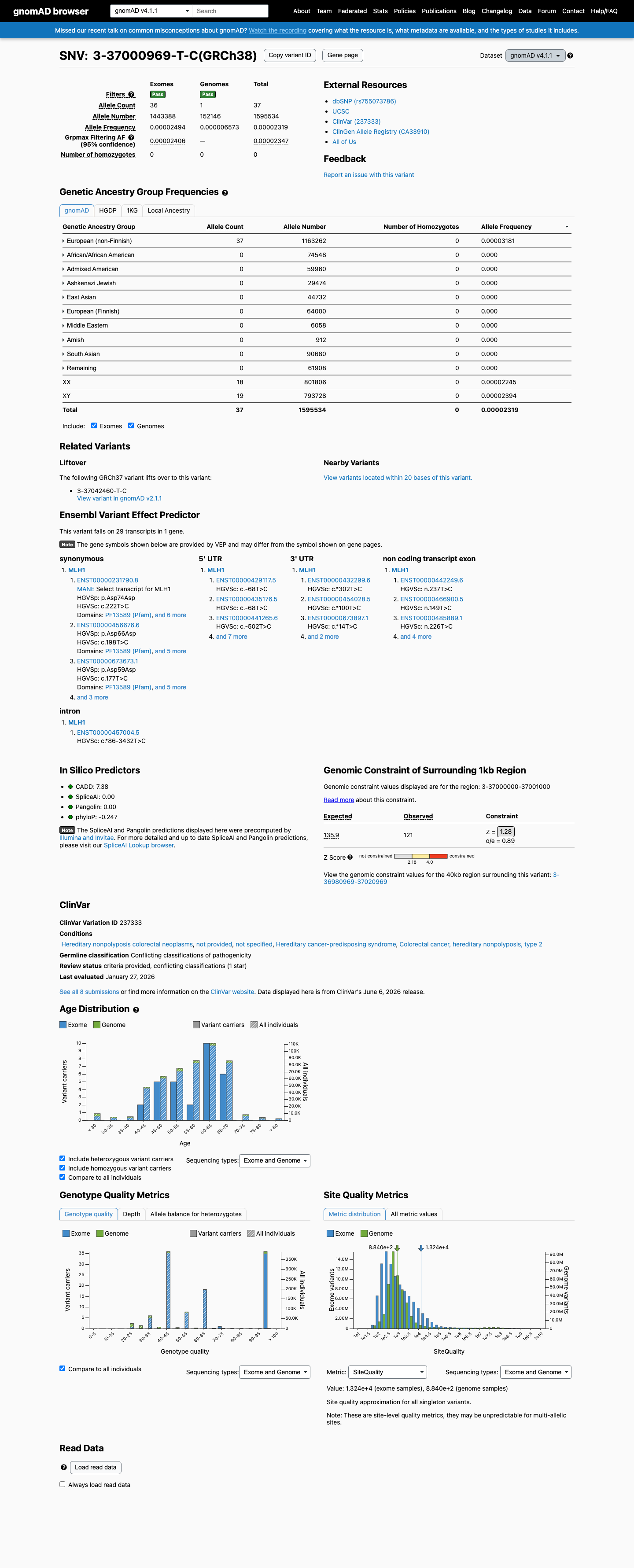
This variant is present in gnomAD v4.1 (AF= 2.31897e-05; MAF= 0.00232%, 37/1595534 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.18071e-05; MAF= 0.00318%, 37/1163262 alleles, homozygotes = 0); grpmax FAF= 2.347e-05.
v2.1
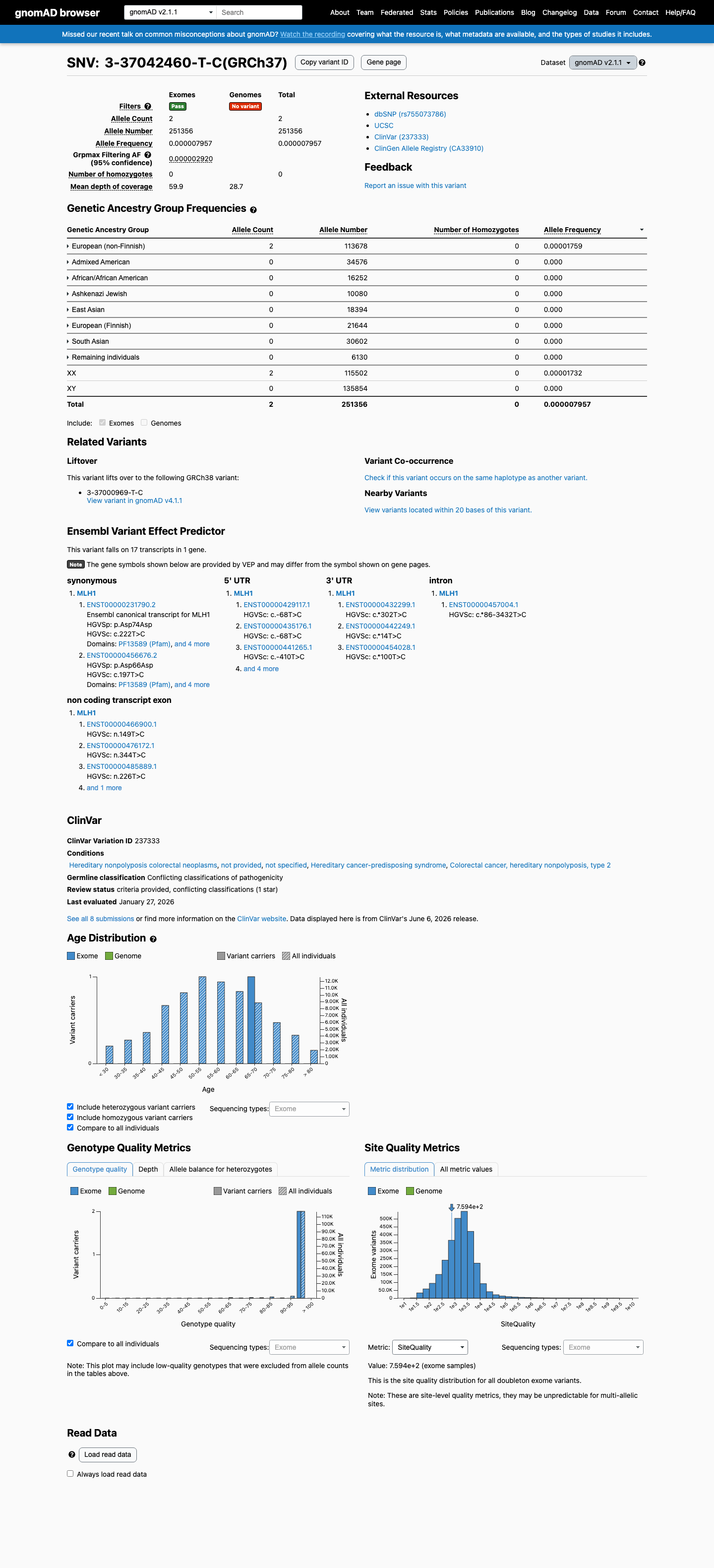
This variant is present in gnomAD v2.1 (AF= 7.95684e-06; MAF= 0.00080%, 2/251356 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.75936e-05; MAF= 0.00176%, 2/113678 alleles, homozygotes = 0); grpmax FAF= 2.92e-06.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0023%
· 37 / 1,595,534
0 hom · FAF 0.0023%
0 hom · FAF 0.0023%
European (non-Finnish) 37 / 1,163,262 |
0.0032% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0008%
· 2 / 251,356
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
European (non-Finnish) 2 / 113,678 |
0.0018% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
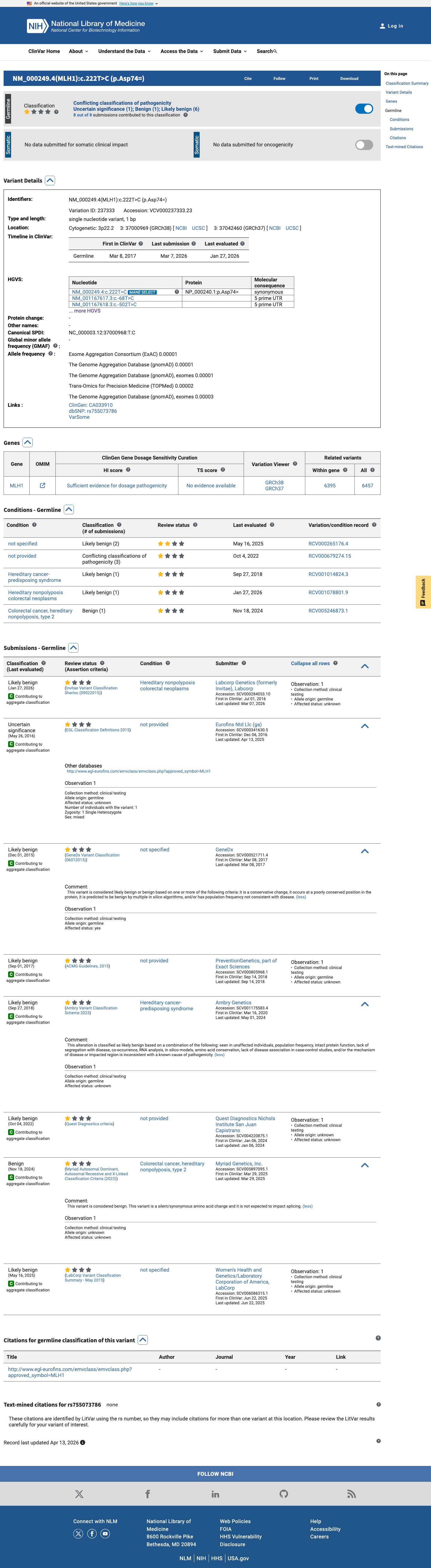
This variant has been reported in ClinVar as Likely benign (6 clinical laboratories) and as Uncertain significance (1 clinical laboratory) and as Benign (1 clinical laboratory). (ClinVarID = 237333)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03). REVEL score = 0.035.
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 7 further PMIDs triaged but not cited — see Sources & References.
ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes.
Found
Structured finding pending for this record — see source link.
Applied to
→BS3 supports · met
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
Found
Structured finding pending for this record — see source link.
Applied to
→BS3 supports · met
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25070057 ↗
Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
11598466 ↗
Practice parameters for the identification and testing of patients at risk for dominantly inherited colorectal cancer--supporting documentation.
CLINVAR
20065170 ↗
American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility.
CLINVAR
22167527 ↗
Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR