Classification rationale
BA1
Benign
MLH1 c.290A>G
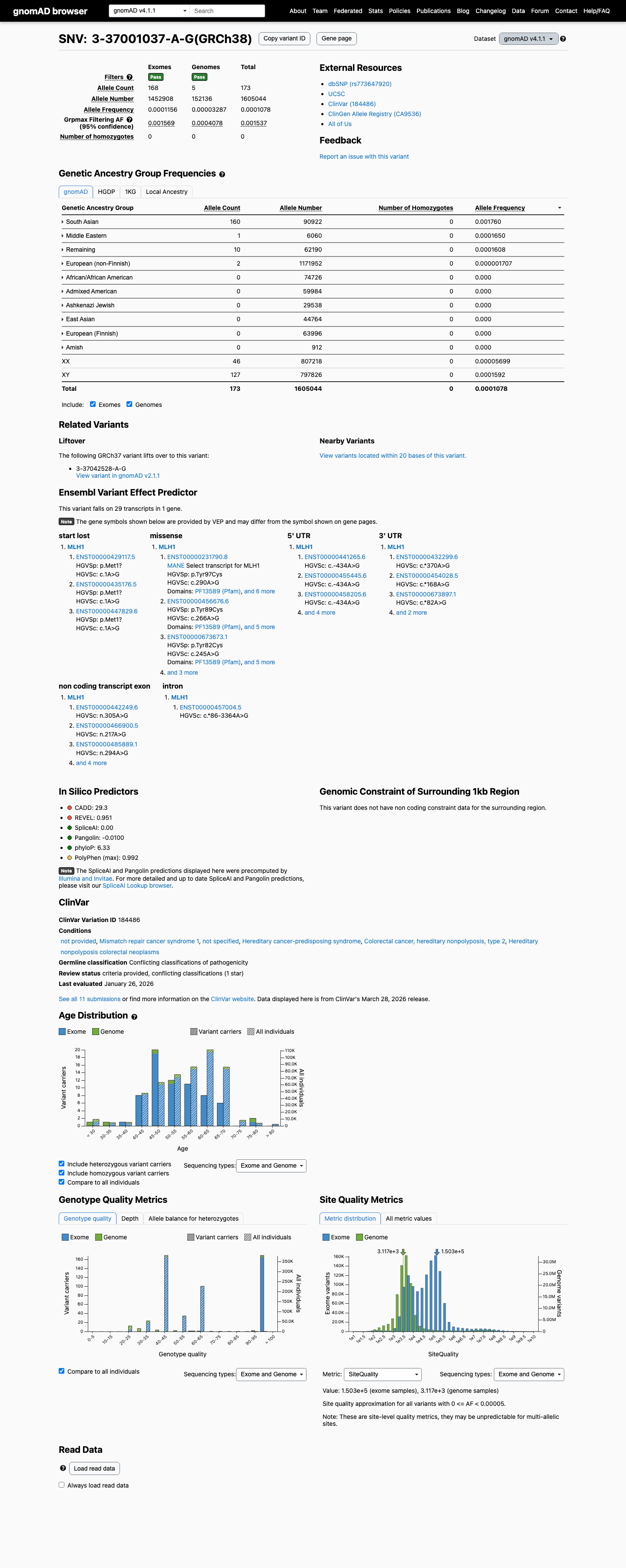
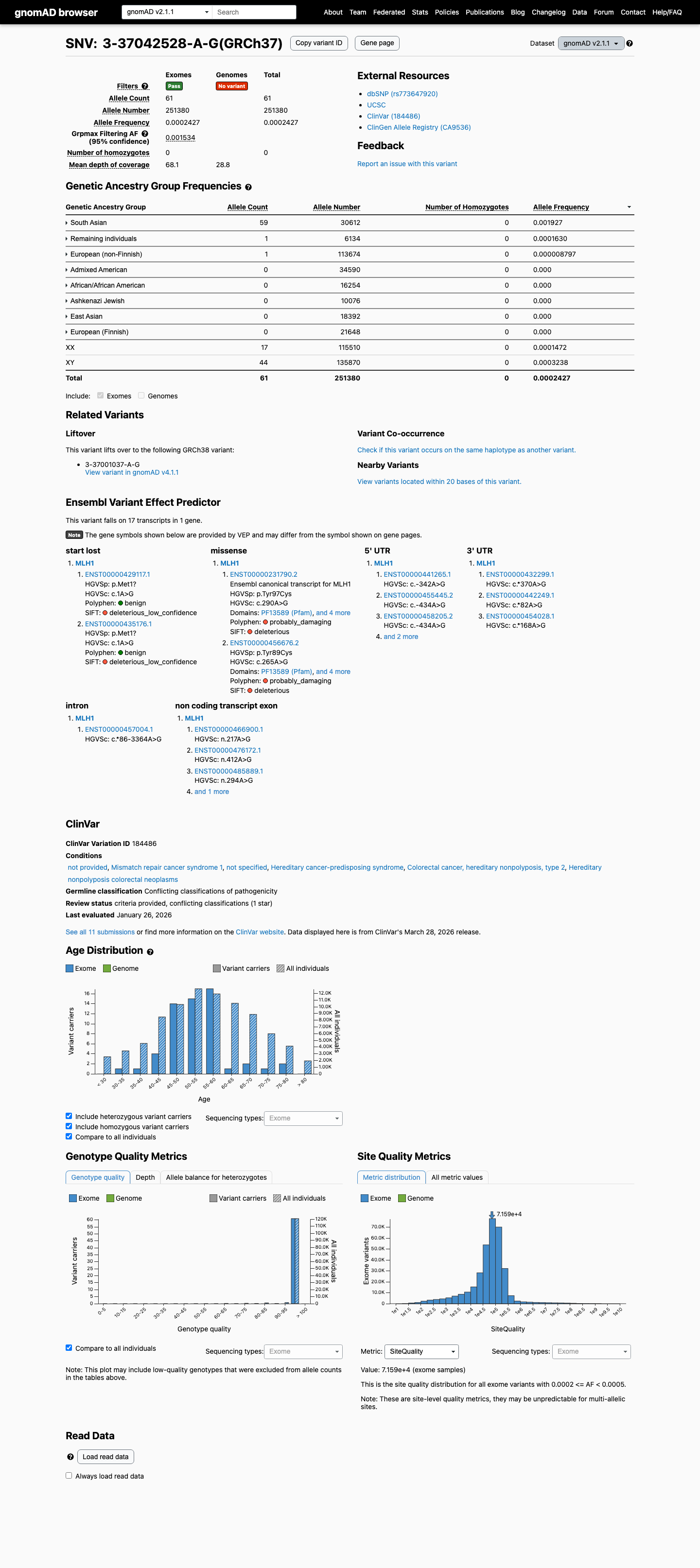
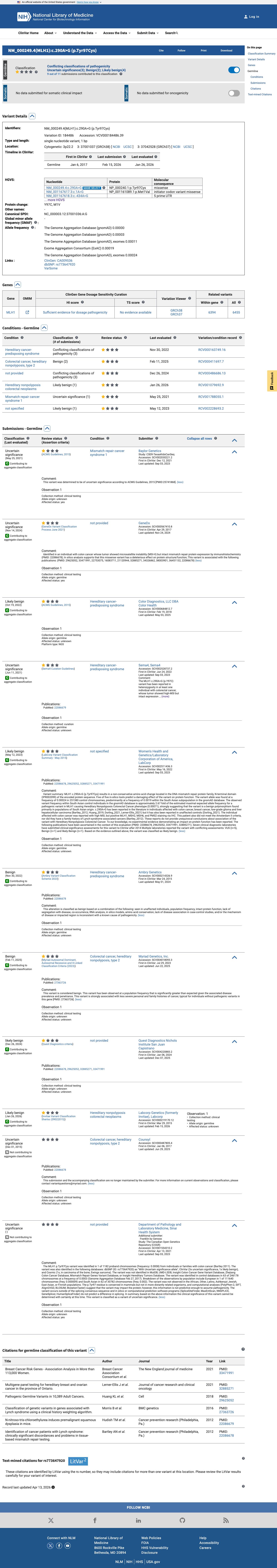
The MLH1 c.290A>G (p.Tyr97Cys) variant has been observed once in somatic cancers in COSMIC and has been reported in ClinVar with conflicting germline interpretations, including uncertain significance, likely benign, and benign submissions.1 This variant is present in gnomAD at a South Asian grpmax filtering allele frequency of 0.0015372 in v4.1, which is above the MLH1 VCEP BA1 threshold of 0.001; gnomAD v2.1 also shows elevated South Asian frequency with grpmax FAF 0.00153368.2 SpliceAI predicts no significant splice impact for this variant with a maximum delta score of 0.01; REVEL is 0.603 and BayesDel is 0.230019, but no MLH1 VCEP HCI prior probability was identified to support PP3 or BP4 for this missense change.3
BA1
→
Benign