NM_000251.2:c.1381G>A (p.Asp461Asn) is a missense variant in MSH2 exon 8. It is absent from ClinVar and extremely rare in gnomAD v4.1 (1/1,584,114 alleles; AF=6.31e-07), meeting PM2_Supporting under the InSiGHT MSH2 VCEP v2.0.0.1 In silico predictions are concordantly benign: HCI prior probability is 0.0014 (<0.11 threshold), REVEL is 0.296, BayesDel is -0.07, and SpliceAI predicts no splicing impact (max delta=0.01). This meets BP4_Supporting under the VCEP framework.2 No functional data, co-segregation data, tumor phenotype data, or de novo observations are available for this variant. No same-residue comparator variants classified as pathogenic by this VCEP were identified. PVS1, PS1, PS2, PS3, PS4, PS5, PM1, PM5, PM6, PP1, PP2, PP3, PP4, PP5, BA1, BS1, BS2, BS3, BS4, BP1, BP2, BP5, BP6, and BP7 are either not met or not applicable.3 With one pathogenic supporting criterion (PM2_Supporting) and one benign supporting criterion (BP4_Supporting), the evidence is insufficient to classify this variant. The variant is classified as a Variant of Uncertain Significance (VUS) under the InSiGHT MSH2 VCEP v2.0.0 combining rules.4
MSH2
Final classification
Uncertain Significance - Conflicting Evidence
MSH2 c.1381G>A · p.Asp461Asn
MSH2
NM_000251.2:c.1381G>A (p.Asp461Asn) is a missense variant in MSH2 exon 8. It is absent from ClinVar and extremely rare in gnomAD v4.1 (1/1,584,114 alleles; AF=6.31e-07), meeting PM2_Supporting under the InSiGHT MSH2 VCEP v2.0.0.
Richards et.al., 2015 - Combining rules v2.0.0 criteria-combination framework: matched Rule31 (Benign.Supporting >=1 + Pathogenic.Supporting >=1) with applied criteria: PM2 supporting, BP4 supporting benign; maps to Uncertain Significance - Conflicting Evidence.
Classification rationale
PM2
BP4
Uncertain Significance - Conflicting Evidence
MSH2 c.1381G>A
PM2 + BP4
→
Uncertain Significance - Conflicting Evidence
Gene diagram
· NM_000251.2 · variants mapped to exon structure
MSH2
NM_000251.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 13 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present in gnomAD v4.1 at an allele frequency of 6.31e-07 (1/1,584,114 alleles; 0 homozygotes), well below the VCEP PM2_Supporting threshold of <0.00002 (<1 in 50,000 alleles). It is absent from gnomAD v2.1.
gnomAD v4.1: AF=6.31e-07 (1/1584114 alleles)
✓
BP4
supporting
Benign
HCI prior probability for pathogenicity is 0.0014, well below the VCEP BP4_Supporting threshold of <0.11. Multiple in silico predictors concordantly suggest a benign effect: REVEL=0.296, BayesDel=-0.07, SpliceAI max delta=0.01 (no splicing impact).
HCI prior=0.0014 (BP4_Supporting threshold: <0.11). REVEL=0.296. BayesDel=-0.07. SpliceAI max delta=0.01 (no splicing alteration).
Assessed · not applied
Pathogenic
PS1
No different nucleotide change encoding the same amino acid substitution (p.Asp461Asn) has been previously classified as Pathogenic or Likely Pathogenic by this VCEP.
PS2
No de novo observations for this variant have been reported in the literature or clinical databases.
PS3
No functional assay data exists for NM_000251.2:c.1381G>A (p.Asp461Asn).
PM5
No different missense change at codon 461 has been classified as Pathogenic or Likely Pathogenic by this VCEP.
PP1
No co-segregation data available for this variant.
PP3
In silico predictions do not support a deleterious effect.
PP4
No tumor data (MSI status, MMR immunohistochemistry) are available to assess whether the patient's phenotype is specific for Lynch syndrome.
Benign
BA1
gnomAD v4.1 allele frequency is 6.31e-07, far below the VCEP BA1 threshold of >=0.001 (>=0.1%).
BS1
gnomAD v4.1 allele frequency is 6.31e-07, far below the VCEP BS1 threshold of >=0.0001 (>=0.01%).
BS2
No data on co-occurrence in trans with a known pathogenic MSH2 variant in an individual without CMMRD features.
BS3
No functional data demonstrating normal or proficient MMR function for this variant.
BS4
No co-segregation data available to assess lack of segregation with disease.
BP5
No tumor data available to assess MSS status or MMR protein expression.
N/A · 13
PVS1 · PS4 · PM1 · PM3 · PM4 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
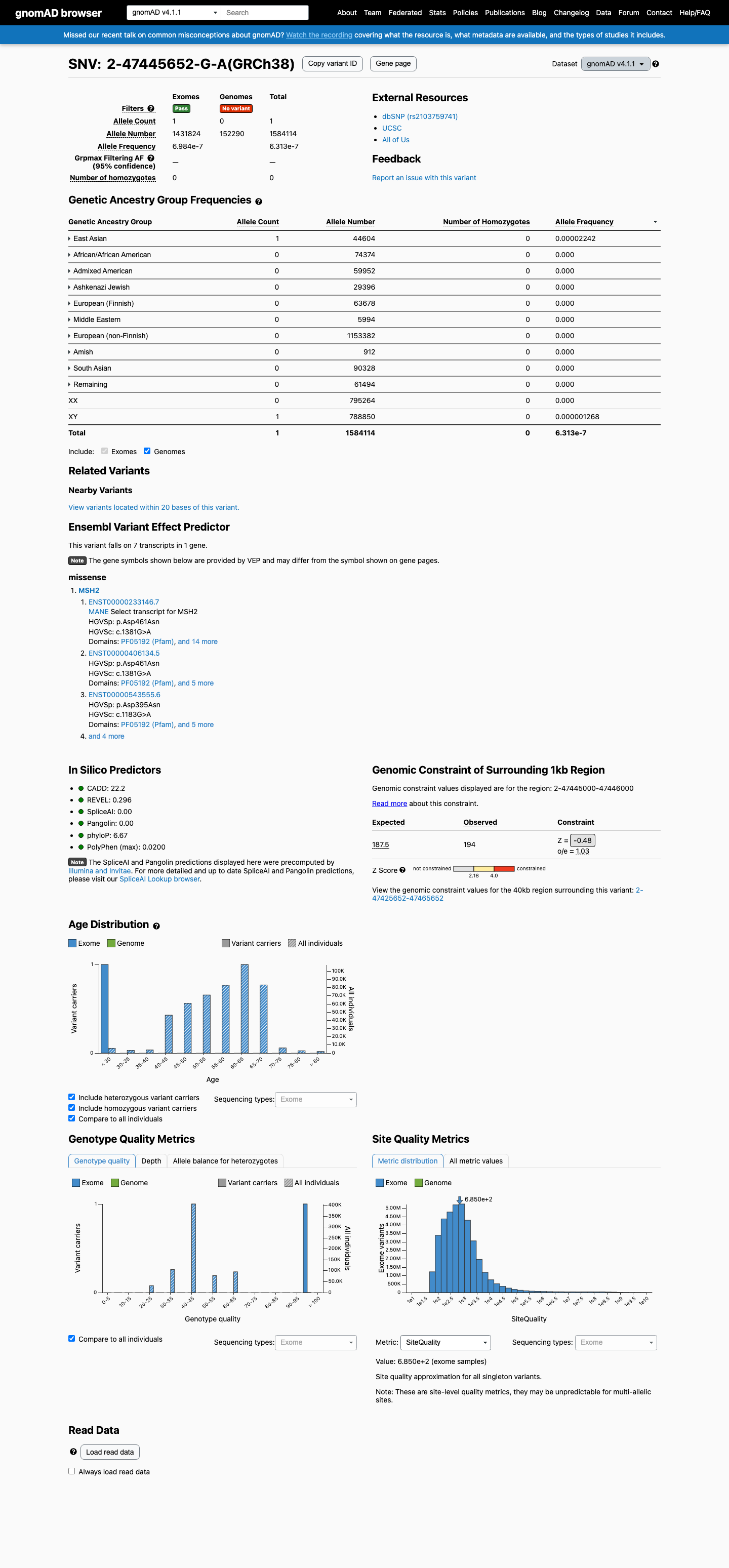
This variant is present in gnomAD v4.1 (AF= 6.31268e-07; MAF= 0.00006%, 1/1584114 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 2.24195e-05; MAF= 0.00224%, 1/44604 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.3e-05%
· 1 / 1,584,114
0 hom
0 hom
East Asian 1 / 44,604 |
0.0022% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

In silico
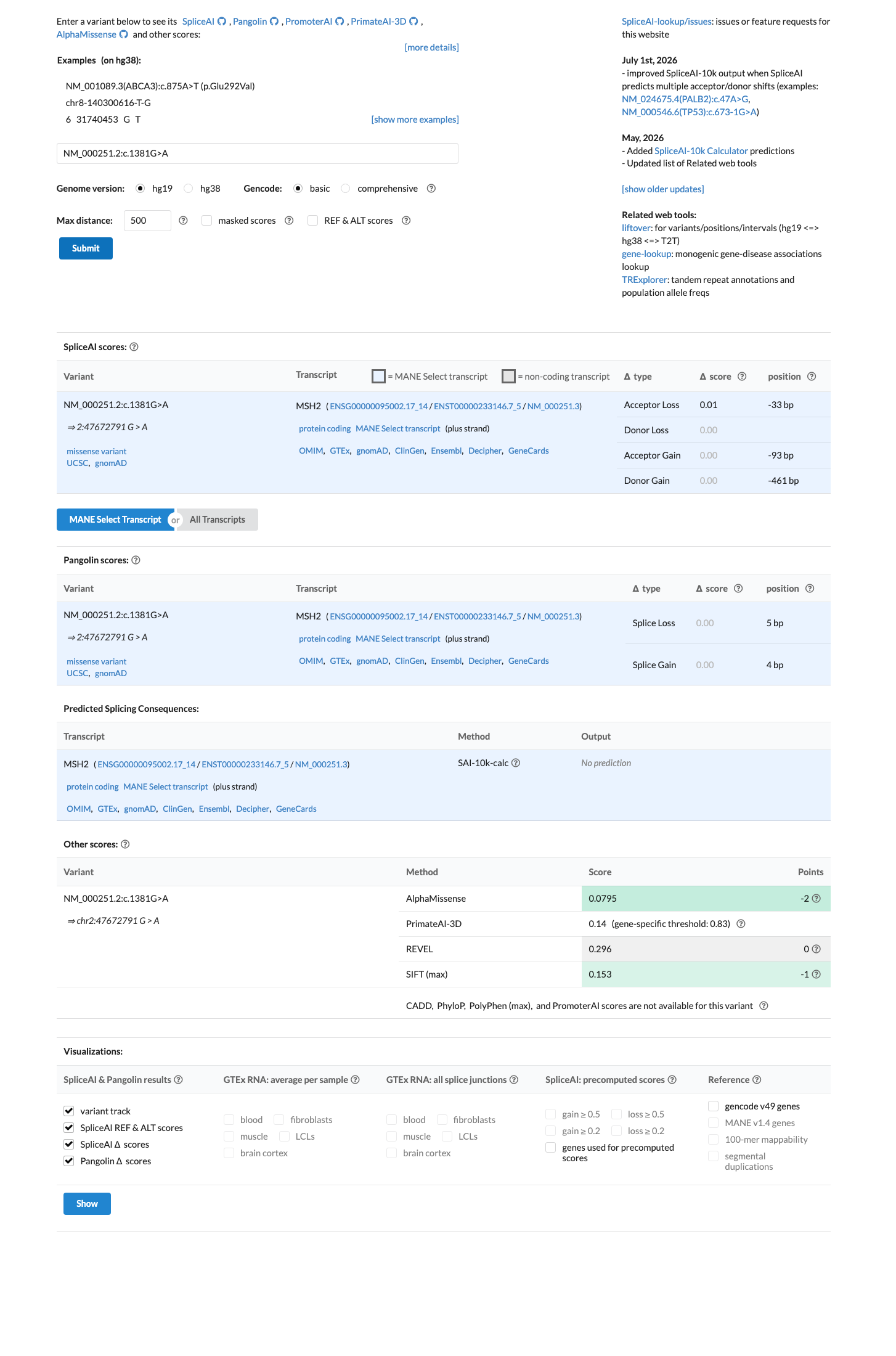
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.296. BayesDel score = -0.0698272. HCI prior probability for pathogenicity = 0.0014. MAPP score = 2.8. Custom PP2 score = 0.069.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MSH2, a DNA mismatch repair protein, is frequently mutated in colorectal, small bowel, and endometrial cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links