PVS1_VeryStrong is met: c.198C>A (p.Tyr66Ter) is a nonsense variant introducing a premature termination codon at codon 66, which is ≤ codon 891 in MSH2, satisfying the InSiGHT MSH2 v2.0.0 VCEP PVS1 rule for full-strength PVS1.1 PM2_Supporting is met: the variant is absent from gnomAD v4.1 (0/1,601,554 alleles) and gnomAD v2.1, satisfying the VCEP PM2_Supporting threshold of <0.00002 (<1 in 50,000 alleles).2 Under the InSiGHT MSH2 v2.0.0 VCEP combination rules (Richards et al. 2015 framework), the combination of 1 Pathogenic Very Strong (PVS1) and 1 Pathogenic Supporting (PM2) does not satisfy any rule for Pathogenic or Likely Pathogenic classification. A minimum of 1 Very Strong + 2 Supporting, or 1 Very Strong + 1 Moderate would be required for Pathogenic or Likely Pathogenic, respectively. The variant is classified as Variant of Uncertain Significance (VUS) per the VCEP combination rule set.3 This variant has been reported as Pathogenic by 6 clinical laboratories in ClinVar (VariationID: 645593). However, the VCEP does not recognize PP5/ClinVar consensus as an applicable criterion, and no expert panel classification has been issued for this variant.4 No variant-specific functional (PS3/BS3), segregation (PP1/BS4), tumor phenotype (PP4/BP5), or de novo (PS2) data were identified for this variant in any reviewed source. Full-text publications retrieved for relevant PMIDs (24362816, 10946232, 11257106, 15528792, 23391514) did not contain accessible article content — all returned Sci-Hub landing page artifacts without variant-specific evidence.
MSH2
Final classification
VUS
MSH2 c.198C>A · p.Tyr66Ter
MSH2
PVS1_VeryStrong is met: c.198C>A (p.Tyr66Ter) is a nonsense variant introducing a premature termination codon at codon 66, which is ≤ codon 891 in MSH2, satisfying the InSiGHT MSH2 v2.0.0 VCEP PVS1 rule for full-strength PVS1.
Richards et.al., 2015 - Combining rules v2.0.0 criteria-combination framework was evaluated deterministically with applied criteria: PVS1 very strong, PM2 supporting; no rule matched the adjudicated criteria.
Classification rationale
PVS1PM2
VUS
MSH2 c.198C>A
PVS1 + PM2
→
VUS
Gene diagram
· NM_000251.3 · variants mapped to exon structure
MSH2
NM_000251.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 10 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
Nonsense variant NM_000251.3:c.198C>A (p.Tyr66Ter) introduces a premature termination codon at codon 66, which is ≤ codon 891 in MSH2, satisfying the VCEP InSiGHT MSH2 v2.0.0 PVS1_VeryStrong rule. Loss of function is an established disease mechanism for MSH2 in Lynch syndrome as confirmed by the ClinGen InSiGHT CSPEC framework.
Nonsense variant at codon 66 (≤891) meets VCEP PVS1_VeryStrong thresholdMSH2 germline LoF established as disease mechanism per CSPEC PVS1 guidancePVS1 decision tree for MMR genes confirms nonsense variants ≤ codon 891 qualify for full-strength PVS1
✓
PM2
supporting
Pathogenic
The variant is absent from gnomAD v4.1 (0/1,601,554 alleles, AF=0.00000%), satisfying the VCEP InSiGHT MSH2 v2.0.0 PM2_Supporting threshold of <0.00002 (<1 in 50,000 alleles). Also absent from gnomAD v2.1 and gnomAD-Canada v1.0.
gnomAD v4.1: 0/1601554 alleles (AF=0.00000%)
Assessed · not applied
Pathogenic
PS2
No de novo data available for this variant.
PS3
No variant-specific calibrated functional assay data available.
PP1
No co-segregation data available for this variant.
PP4
No tumor phenotype data available for this variant.
Benign
BA1
VCEP InSiGHT MSH2 v2.0.0 BA1 requires gnomAD v4 Grpmax filtering allele frequency ≥0.001 (0.1%).
BS1
VCEP InSiGHT MSH2 v2.0.0 BS1 requires gnomAD v4 Grpmax filtering allele frequency ≥0.0001 and <0.001 (0.01-0.1%).
BS2
No data available for co-occurrence in trans with a known pathogenic MSH2 variant.
BS3
No variant-specific functional data demonstrating proficient MMR function.
BS4
No segregation data available.
BP5
No tumor phenotype data available.
N/A · 13
PS1 · PS4 · PM1 · PM5 · PM6 · PP2 · PP3 · PP5 · BP1 · BP2 · BP4 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
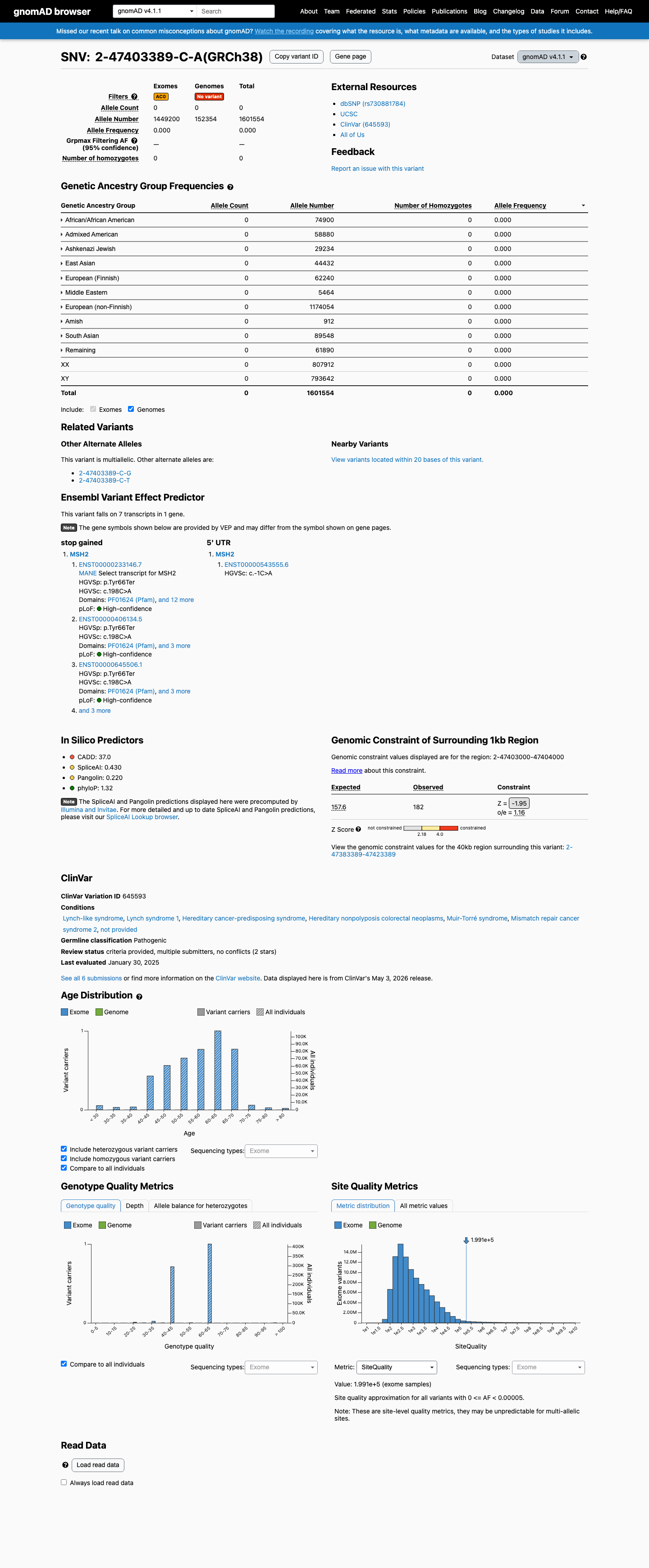
This variant is present in gnomAD v4.1 (AF= 0; MAF= 0.00000%, 0/1601554 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/74900 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / 1,601,554
0 hom
0 hom
Not observed in any ancestry group.
+ 10 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
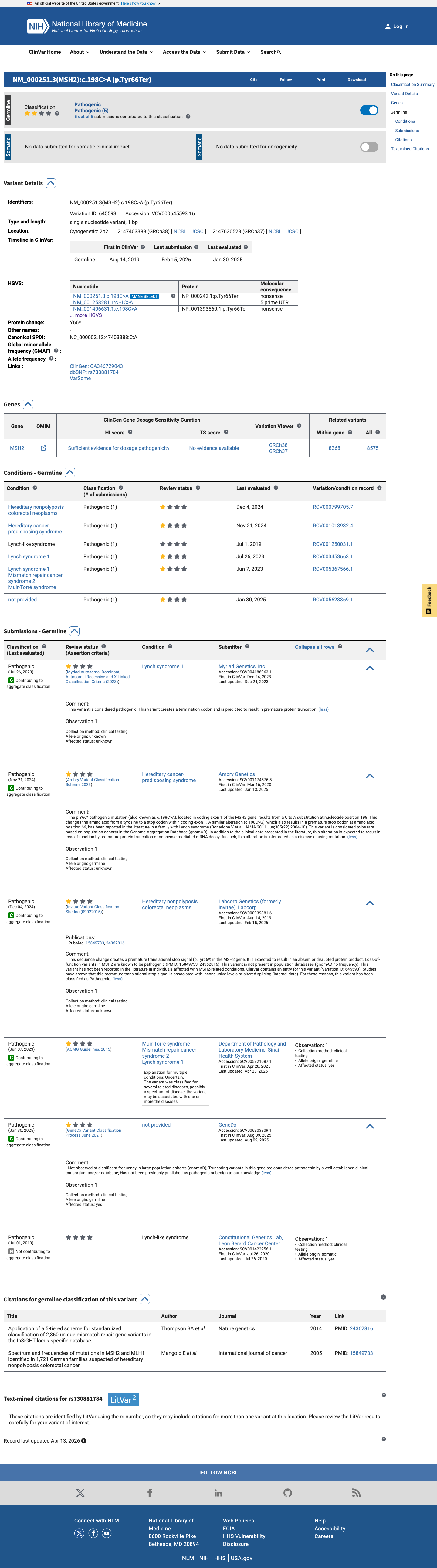
This variant has been reported in ClinVar as Pathogenic (6 clinical laboratories). (ClinVarID = 645593)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). BayesDel score = 0.66.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
24362816 ↗
Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database.
ONCOKB
15528792 ↗
Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database.
ONCOKB
23391514 ↗
Structural, molecular and cellular functions of MSH2 and MSH6 during DNA mismatch repair, damage signaling and other noncanonical activities.
ONCOKB
7726159 ↗
Seven new mutations in hMSH2, an HNPCC gene, identified by denaturing gradient-gel electrophoresis.
ONCOKB
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR