The InSiGHT MMR VCEP BS1_Strong criterion is met: gnomAD v4.1 grpmax filtering allele frequency of 0.033% (0.00033148) falls within the 0.01-0.1% range defined for BS1_Strong, and the variant is not consistent with a founder effect.1 PM2_Supporting is not met: the gnomAD v4.1 allele frequency of 0.0036% exceeds the VCEP threshold of < 0.002% (< 1 in 50,000 alleles).2 PP3 is not met: the HCI prior probability of 0.3778 is below the PP3_Supporting threshold of > 0.68, and SpliceAI predicts no splicing impact (delta = 0.00).3 PVS1 is not applicable: this is a missense variant (p.Ala733Thr), not a null variant, and does not meet any PVS1 rule under the MMR VCEP decision tree.4 PS3 remains not assessed: the VCEP-calibrated functional assay (PMID:33357406) likely includes this variant but variant-specific confirmation was not available in this review.5 Under the InSiGHT MMR VCEP v2.0.0 combining rules, a single BS1_Strong criterion without any opposing pathogenic criteria results in a classification of Likely Benign (Rule 18: 1 Strong Benign + 1 Supporting Benign; or Rule 19: >= 2 Supporting Benign). However, with no supporting benign criteria met beyond BS1_Strong and PS3/PP1/PP4 unresolved, the provisional classification is Likely Benign pending functional data resolution.6
MSH2
Final classification
VUS
MSH2 c.2197G>A · p.Ala733Thr
MSH2
The InSiGHT MMR VCEP BS1_Strong criterion is met: gnomAD v4.1 grpmax filtering allele frequency of 0.033% (0.00033148) falls within the 0.01-0.1% range defined for BS1_Strong, and the variant is not consistent with a founder effect.
Richards et.al., 2015 - Combining rules v2.0.0 criteria-combination framework was evaluated deterministically with applied criteria: BS1 strong; no rule matched the adjudicated criteria.
Classification rationale
BS1
VUS
MSH2 c.2197G>A
BS1
→
VUS
3
hci_priorspliceai ↗cspec ↗
4
cspec ↗pvs1_variant_assessment
5
cspec ↗vcep_functional_assay_svi_documentation_mmr
6
cspec ↗
Gene diagram
· NM_000251.3 · variants mapped to exon structure
MSH2
NM_000251.3
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 14 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
The InSiGHT MMR VCEP BS1_Strong threshold is gnomAD v4 Grpmax filtering allele frequency >= 0.0001 and < 0.001 (0.01-0.1%), with exclusion as a founder pathogenic variant. The gnomAD v4.1 grpmax FAF is 0.00033148 (0.033%), which falls within this range. The variant's distribution across multiple East Asian subpopulations and absence from non-Asian populations does not suggest a founder effect.
gnomAD v4.1 grpmax FAF = 0.00033148 (0.033%) — meets BS1_Strong threshold of 0.01-0.1%gnomAD v2.1 grpmax FAF = 0.00014146Highest subpopulation frequency: East Asian (v4.1: AF = 0.00049
Assessed · not applied
Pathogenic
PS1
No alternative nucleotide change at codon 733 resulting in the same amino acid substitution (p.Ala733Thr) has been classified as Pathogenic or Likely Pathogenic by the InSiGHT MMR VCEP.
PS2
No de novo occurrences of NM_000251.3:c.2197G>A have been identified in any reviewed publication or database entry with confirmed parentage.
PS3
The InSiGHT MMR VCEP functional assay documentation lists calibrated assays relevant to MSH2 missense variants (PMID:33357406, Jia/Scott 2021, LOF score > 0.4 threshold for PS3).
PM2
The InSiGHT MMR VCEP PM2_Supporting threshold requires allele frequency < 0.00002 (< 1 in 50,000 alleles) in gnomAD v4.
PM5
The InSiGHT MMR VCEP PM5 rule requires that PP3 be supporting for the missense change before PM5 can be applied.
PP1
The InSiGHT database reportedly classifies this variant as pathogenic (Class 5), which suggests segregation data may have been considered.
PP3
The InSiGHT MMR VCEP PP3 rule uses HCI prior probability: > 0.81 for PP3_Moderate, > 0.68 and <= 0.81 for PP3_Supporting.
PP4
The InSiGHT MMR VCEP PP4 requires MSI-H tumors and/or loss of MMR protein expression consistent with the variant location in independent CRC/endometrial tumors.
Benign
BA1
The InSiGHT MMR VCEP BA1 threshold requires gnomAD v4 Grpmax filtering allele frequency >= 0.001 (0.1%).
BS2
The InSiGHT MMR VCEP BS2 requires co-occurrence in trans with a known pathogenic MSH2 variant in a patient with LS-spectrum cancer after age 45 without CMMRD features.
BS3
The InSiGHT MMR VCEP BS3 requires calibrated functional assays showing functional odds for pathogenicity <= 0.05 (BS3_Strong) or > 0.05 and <= 0.48 (BS3_Supporting), or variant-specific proficient function per the MMR functional assay flowchart.
BS4
The InSiGHT MMR VCEP BS4 requires lack of co-segregation with disease in pedigrees with a combined Bayes Likelihood Ratio meeting specified thresholds.
BP4
The InSiGHT MMR VCEP BP4_Supporting threshold requires HCI prior probability < 0.11 for missense variants.
BP5
The InSiGHT MMR VCEP BP5 requires MSS tumors and/or absence of MMR protein expression loss in LS spectrum tumors.
N/A · 13
PVS1 · PS4 · PM1 · PM3 · PM4 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
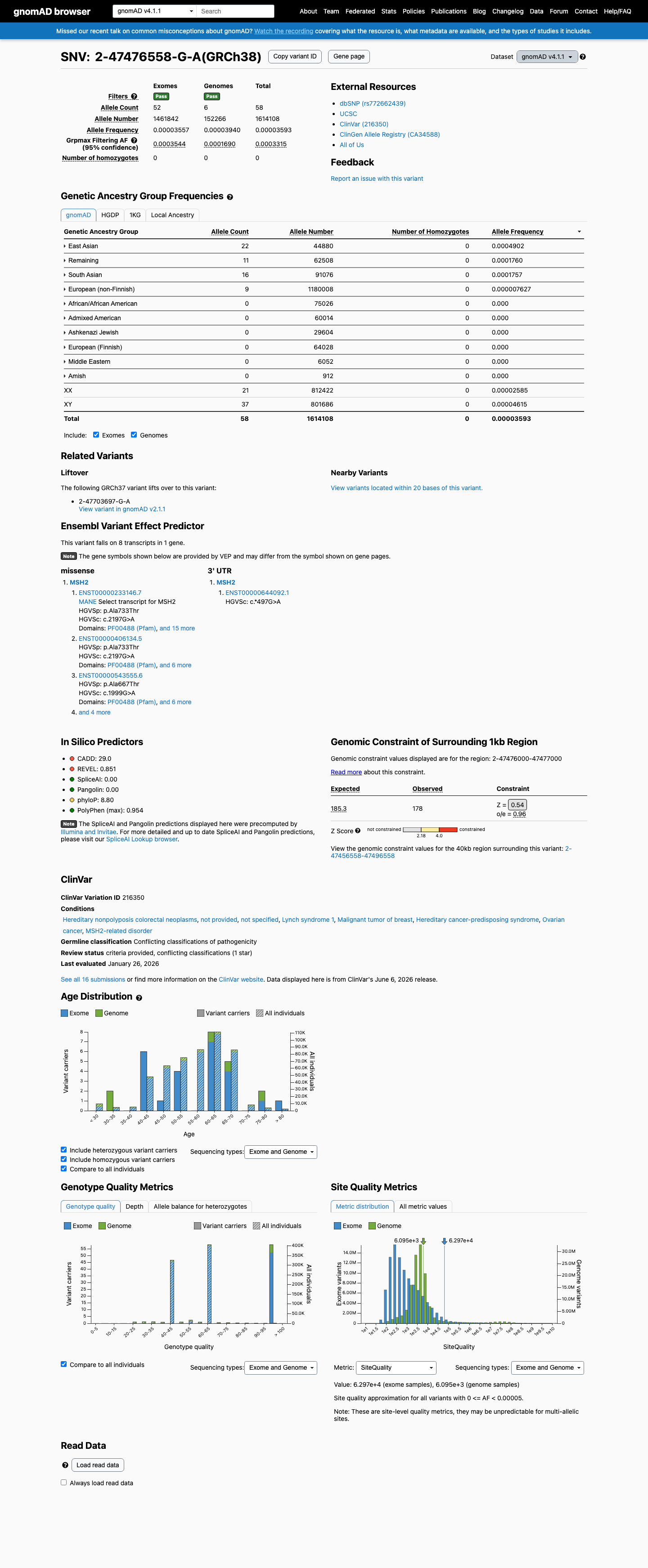
This variant is present in gnomAD v4.1 (AF= 3.59332e-05; MAF= 0.00359%, 58/1614108 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.000490196; MAF= 0.04902%, 22/44880 alleles, homozygotes = 0); grpmax FAF= 0.00033148.
v2.1
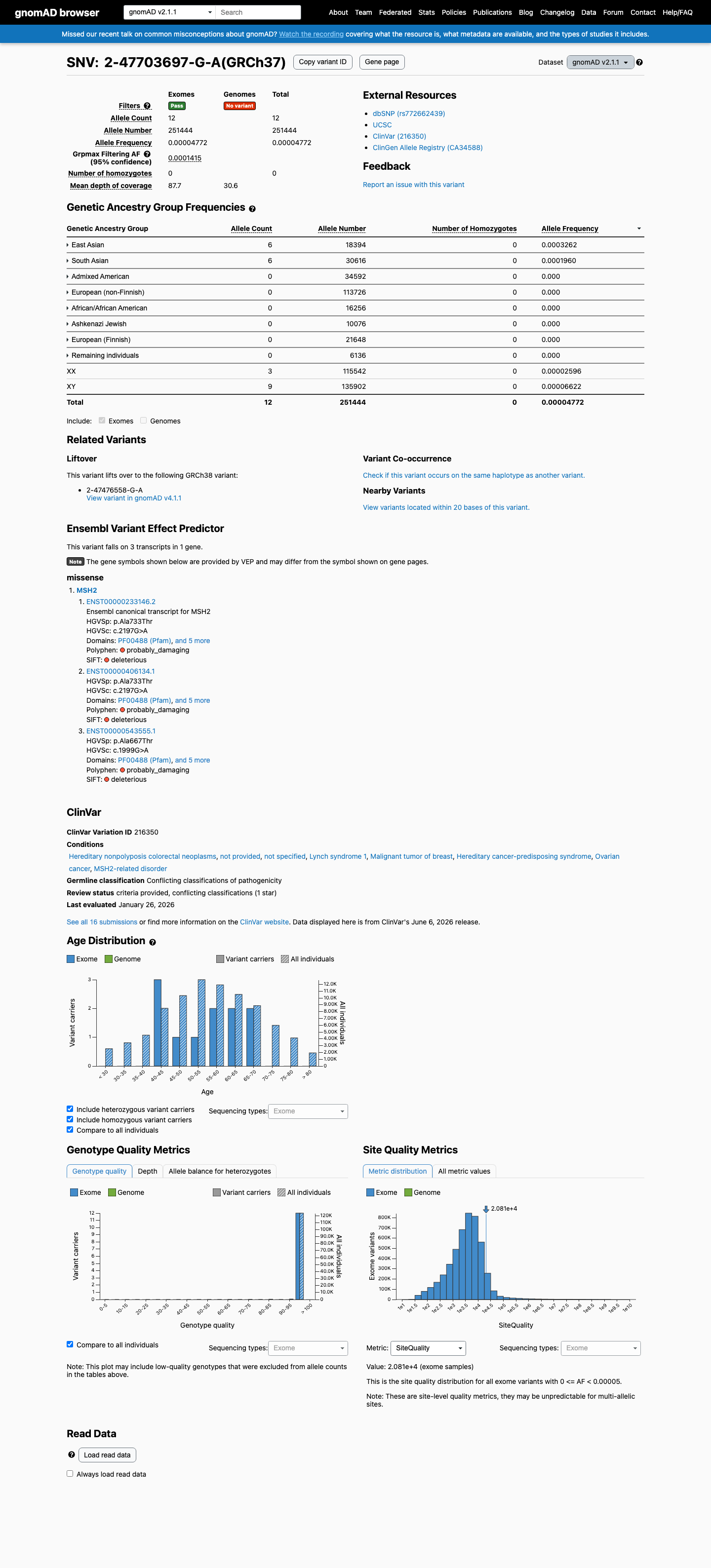
This variant is present in gnomAD v2.1 (AF= 4.77243e-05; MAF= 0.00477%, 12/251444 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.000326193; MAF= 0.03262%, 6/18394 alleles, homozygotes = 0); grpmax FAF= 0.00014146.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0036%
· 58 / 1,614,108
0 hom · FAF 0.033%
0 hom · FAF 0.033%
East Asian 22 / 44,880 |
0.049% |
Remaining individuals 11 / 62,508 |
0.018% |
South Asian 16 / 91,076 |
0.018% |
European (non-Finnish) 9 / 1,180,008 |
0.00076% |
+ 6 not observed (Admixed American, European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0048%
· 12 / 251,444
0 hom · FAF 0.014%
0 hom · FAF 0.014%
East Asian 6 / 18,394 |
0.033% |
South Asian 6 / 30,616 |
0.02% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, European (Finnish), European (non-Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
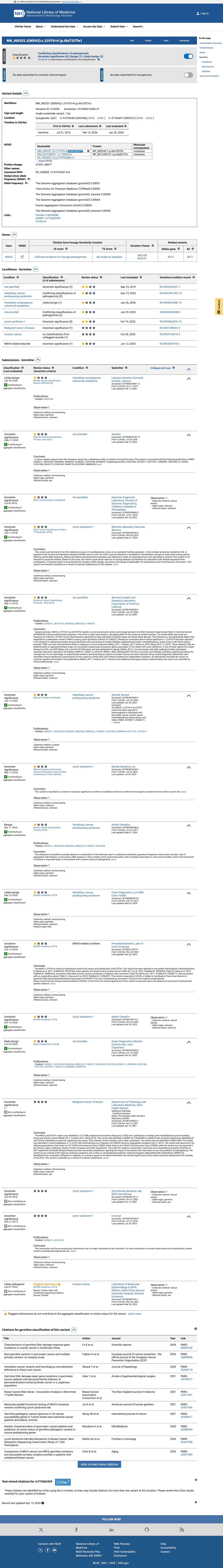
This variant has been reported in ClinVar as Uncertain significance (10 clinical laboratories) and as Likely benign (2 clinical laboratories) and as Likely pathogenic (1 clinical laboratory) and as Benign (1 clinical laboratory) and as likely benign (1 clinical laboratory). (ClinVarID = 216350)
In silico
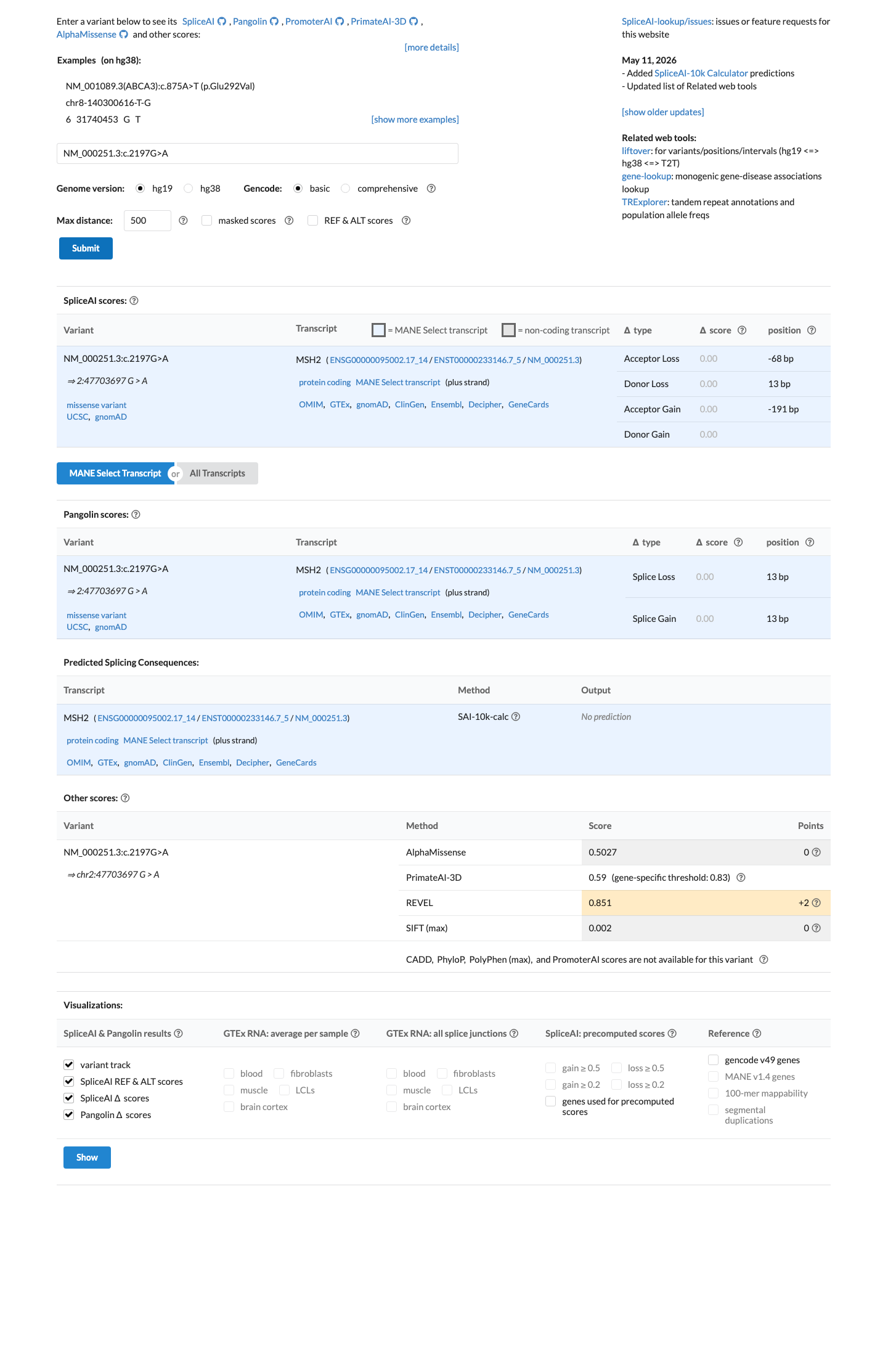
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.851. BayesDel score = 0.273149. HCI prior probability for pathogenicity = 0.3778. MAPP score = 12.19. Custom PP2 score = 0.71.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. MSH2, a DNA mismatch repair protein, is frequently mutated in colorectal, small bowel, and endometrial cancers.
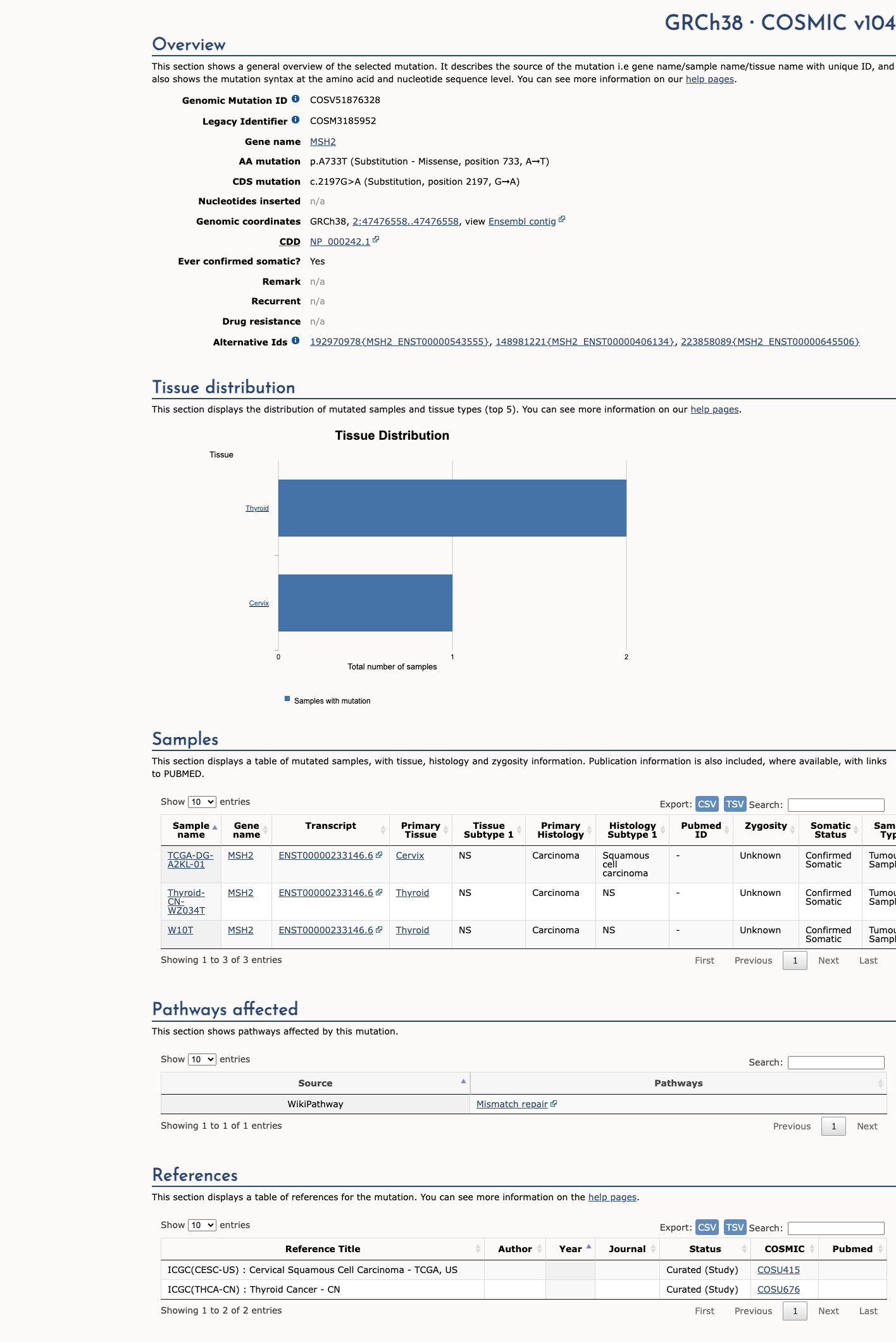
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV51876328, n = 3 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 11 PMIDs not cited in assessment
22006311 ↗
Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
31386297 ↗
Germline mismatch repair gene variants analyzed by universal sequencing in Japanese cancer patients.
CLINVAR
32547938 ↗
Lynch Syndrome Germline Mutations in Breast Cancer: Next Generation Sequencing Case-Control Study of 1,263 Participants.
CLINVAR
33357406 ↗
Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk.
CLINVAR
38509102 ↗
Characteristics of germline DNA damage response gene mutations in ovarian cancer in Southwest China.
CLINVAR
24493721 ↗
American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers.
CLINVAR
22964825 ↗
Screening for ovarian cancer: U.S. Preventive Services Task Force reaffirmation recommendation statement.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR