Classification rationale
BA1
Benign
MSH2 c.67T>C
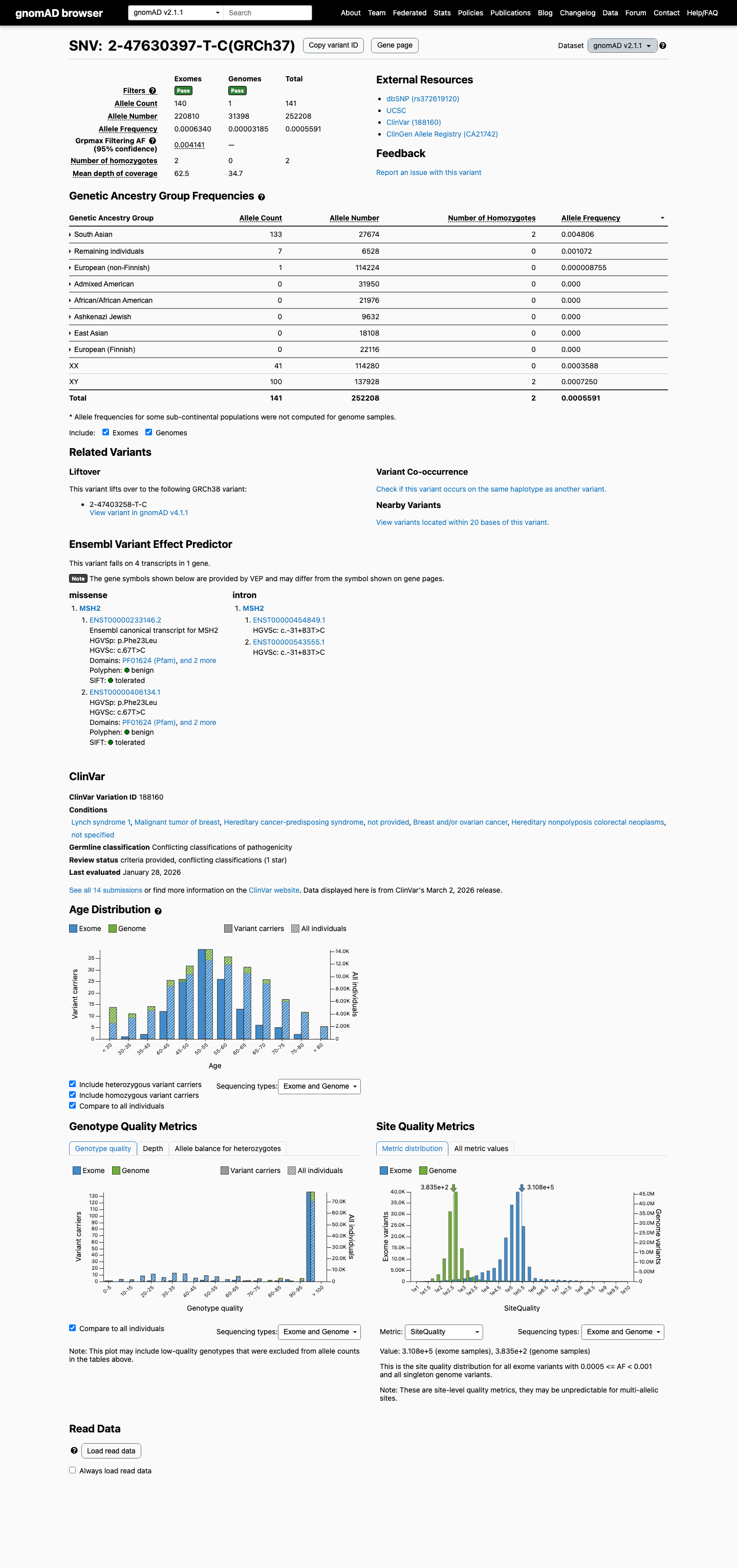
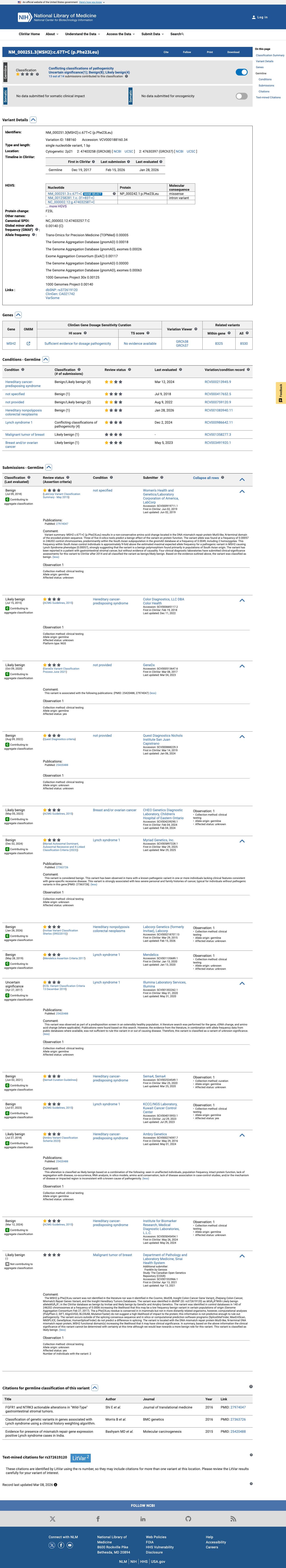
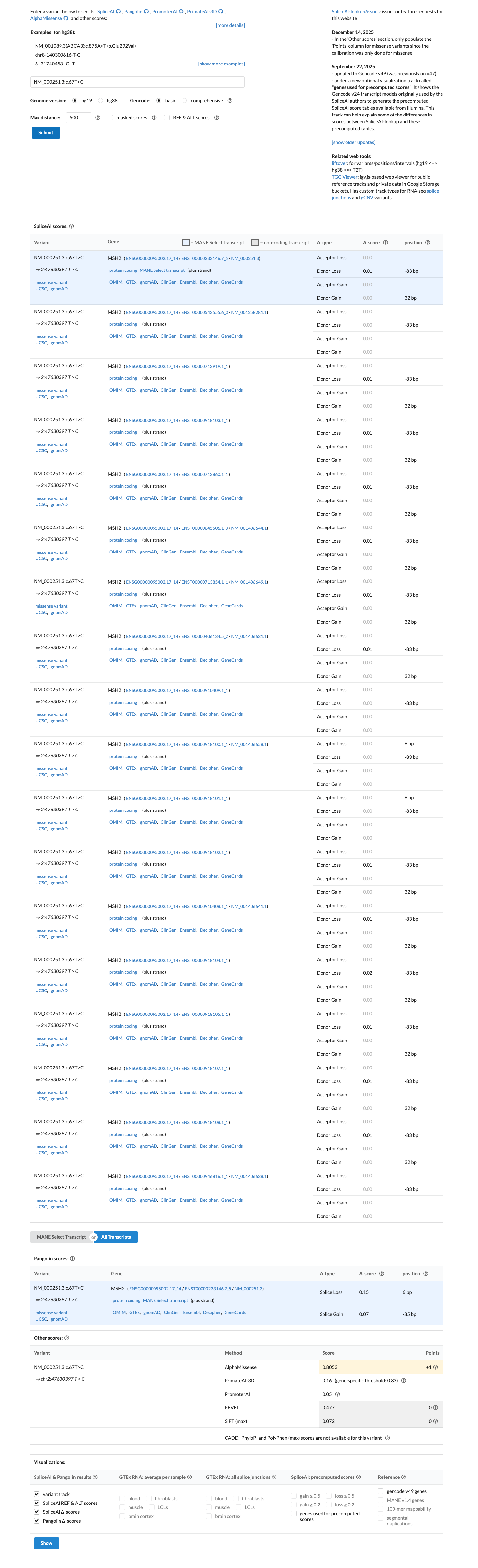
The MSH2 c.67T>C (p.Phe23Leu) variant has not been observed in COSMIC somatic cancer records and has been reported in ClinVar predominantly as benign or likely benign, with an aggregate ClinVar classification of Benign.1 This variant is present at high frequency in population databases, including gnomAD v4.1 at 410/1600398 alleles with 8 homozygotes and grpmax filtering allele frequency 0.00394201, which exceeds the MSH2 VCEP BA1 threshold.2 SpliceAI predicts no significant splice impact for this variant, with a maximum delta score of 0.01; no missense-specific HCI prior value was available to support PP3 or BP4 assessment.3
BA1
→
Benign