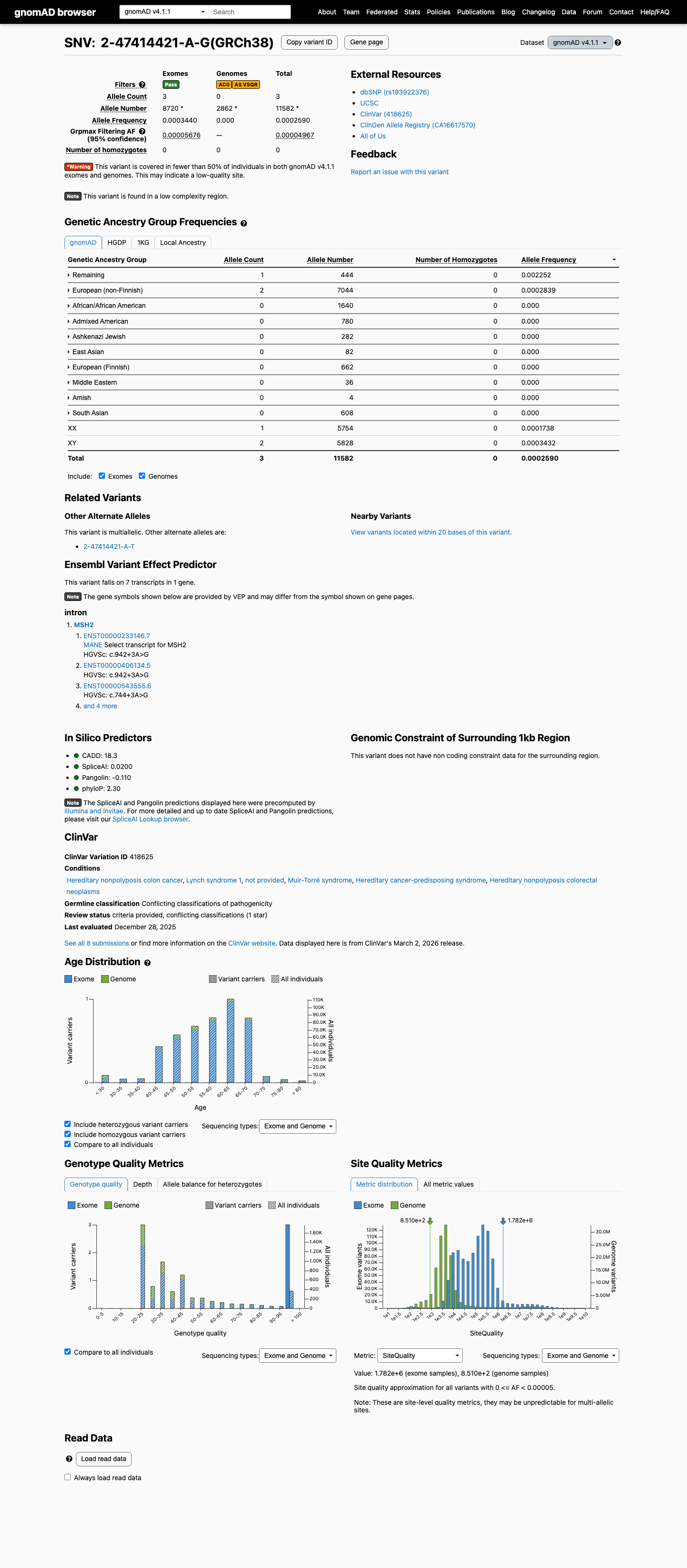
The MSH2 c.942+3A>G (p.?) variant has not been observed in somatic cancers in COSMIC and has been reported in ClinVar with predominantly likely pathogenic submissions, although some submissions classify it as uncertain significance.1 This variant is absent from gnomAD v2.1 but is present in gnomAD v4.1 at 3/11,582 alleles (total AF 0.00025902) with grpmax FAF 0.00004967, which is above the MSH2 VCEP PM2 threshold of less than 0.00002 and below the BS1 threshold of at least 0.0001.2 Published RNA studies reported that this variant disrupts MSH2 donor splicing and generates an exon 5-skipped transcript, supporting a true splice defect despite its non-canonical +3 position.3 SpliceAI predicts a low splice effect with a maximum delta score of 0.02, which is below the MSH2 VCEP PP3 threshold of at least 0.2 and within the BP4 threshold of 0.1 or less, but this in silico result is inconsistent with the published RNA evidence.4
MSH2
Final classification
Uncertain Significance
MSH2 c.942+3A>G · p.?
MSH2
The MSH2 c.942+3A>G (p.?) variant has not been observed in somatic cancers in COSMIC and has been reported in ClinVar with predominantly likely pathogenic submissions, although some submissions classify it as uncertain significance.
Official CSPEC/VCEP final-classification framework from final_classification_framework.json (cspec_ruleset criteria-combination framework; Richards et al. 2015 combining rules as represented in the MSH2/InSiGHT specification).
Classification rationale
PVS1
Uncertain Significance
MSH2 c.942+3A>G
PVS1
→
Uncertain Significance
Gene diagram
· NM_000251.3 · variants mapped to exon structure
MSH2
NM_000251.3
Fetching transcript structure from UCSC…
Applied criteria · 1 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000259023; MAF= 0.02590%, 3/11582 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 0.00225225; MAF= 0.22523%, 1/444 alleles, homozygotes = 0); grpmax FAF= 4.967e-05.
v2.1
Absent from gnomAD v2.1.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.026%
· 3 / 11,582
0 hom · FAF 0.005%
0 hom · FAF 0.005%
Remaining individuals 1 / 444 |
0.23% |
European (non-Finnish) 2 / 7,044 |
0.028% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
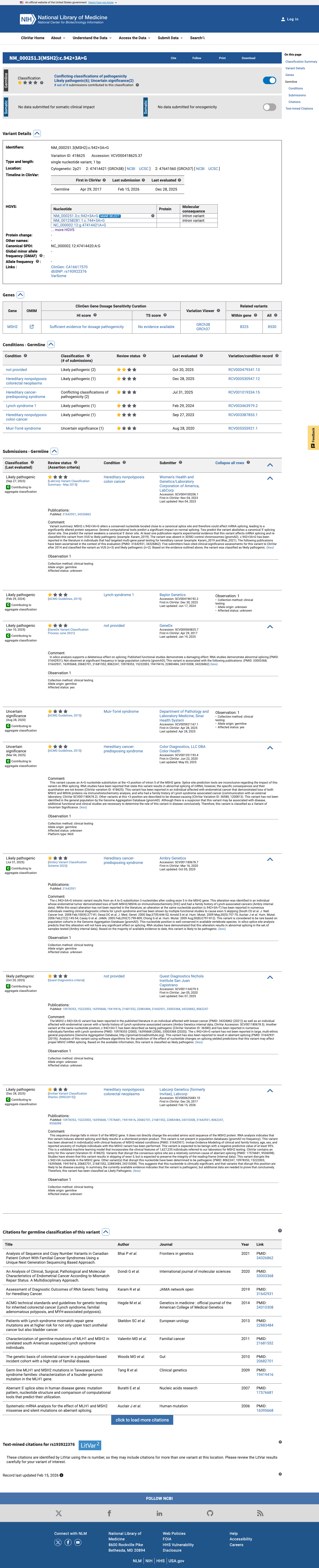
ClinVar
This variant has been reported in ClinVar as Likely pathogenic (5 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as likely pathogenic (1 clinical laboratory).
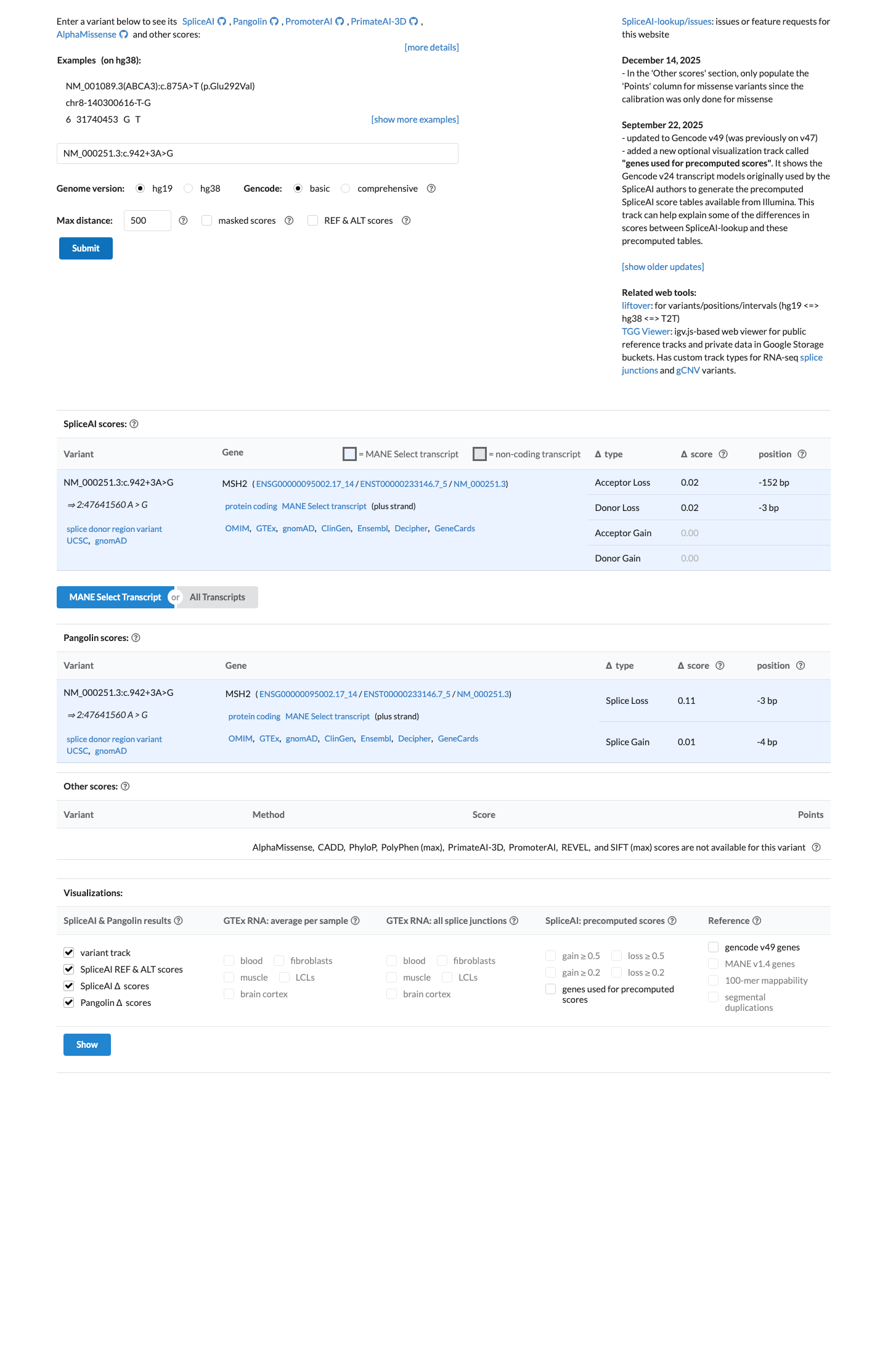
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
Recurrent germline mutation in MSH2 arises frequently de novo.
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
Systematic mRNA analysis for the effect of MLH1 and MSH2 missense and silent mut
Found
Structured finding pending for this record — see source link.
Applied to
→PVS1 supports · met
Sources & reference links