NM_000257.3:c.2548G>A (p.Ala850Thr) is a missense variant in MYH7 exon 22. It is extremely rare in population databases (gnomAD v2.1 AF 3.98e-6, v4.1 grpmax FAF 7.9e-7), satisfying VCEP PM2 at supporting strength.1 Codon 850 falls within the VCEP-defined MYH7 missense cluster region (codons 167-931), meeting PM1 at moderate strength per Walsh et al. 2019 (PMID:30696458).2 REVEL in silico meta-predictor score is 0.809, exceeding the VCEP PP3 threshold of ≥0.70, providing supporting evidence for a deleterious effect. SpliceAI predicts no splice impact.3 A sister missense variant at the same codon, p.Ala850Asp, was reported as a novel mutation in one HCM patient by Fokstuen et al. 2008 (PMID:18409188), but lacks formal VCEP classification necessary for PM5 application.4 No functional studies, segregation data, de novo observations, or case-control data were identified for this specific variant in any reviewed publication. The variant has been reported in ClinVar (ID 403207) as Uncertain Significance by 5 clinical laboratories and Likely Pathogenic by 1 (Invitae). No expert panel classification has been assigned. ClinVar review status is 'criteria provided, single submitter' at the aggregate level.5 Applying the MYH7 VCEP v2.0 classification rules: 1 moderate criterion (PM1) and 2 supporting criteria (PM2_Supporting, PP3) are met. This combination does not satisfy any VCEP Likely Pathogenic rule (minimum: Rule 14 requires 2 moderate + ≥2 supporting; Rule 15 requires 1 moderate + ≥4 supporting). The variant remains a Variant of Uncertain Significance.6
MYH7
Final classification
VUS
MYH7 c.2548G>A · p.Ala850Thr
MYH7
NM_000257.3:c.2548G>A (p.Ala850Thr) is a missense variant in MYH7 exon 22. It is extremely rare in population databases (gnomAD v2.1 AF 3.98e-6, v4.1 grpmax FAF 7.9e-7), satisfying VCEP PM2 at supporting strength.
ClinGen Cardiomyopathy Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for MYH7 Version 2.0 v2.0 criteria-combination framework was evaluated deterministically with applied criteria: PM1 moderate, PM2 supporting, PP3 supporting; no rule matched the adjudicated criteria.
Classification rationale
PM1PM2PP3
VUS
MYH7 c.2548G>A
PM1 + PM2 + PP3
→
VUS
Gene diagram
· NM_000257.3 · variants mapped to exon structure
MYH7
NM_000257.3
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 13 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
Codon 850 lies within the VCEP-defined MYH7 missense cluster region (codons 167-931) per Walsh et al. 2019 (PMID:30696458). This region shows significant enrichment of rare missense variants in HCM case cohorts.
VCEP MYH7 v2.0 PM1 specification: codons 167-931 eligibleCodon 850 falls within the defined cluster region.
✓
PM2
supporting
Pathogenic
Variant is extremely rare in population databases. gnomAD v2.1 NFE AF = 8.79e-6 (1/113,748 alleles), well below the VCEP PM2 threshold of ≤0.00004. Absent from Ashkenazi Jewish and Other subpopulations. gnomAD v4.1 grpmax FAF = 7.9e-7.
gnomAD v2.1: 1/251470 total alleles (AF 3.98e-6)gnomAD v2.1 NFE: 1/113
✓
PP3
supporting
Pathogenic
REVEL score 0.809 exceeds the VCEP-recommended threshold of ≥0.70 for PP3 application. SpliceAI predicts no splice impact (max delta = 0.00), so the in silico signal is confined to missense pathogenicity prediction.
REVEL score: 0.809 (≥0.70 threshold met)SpliceAI max delta: 0.00 (no splice impact)
Assessed · not applied
Pathogenic
PS1
No evidence of a different nucleotide change at codon 850 producing the same amino acid change (p.Ala850Thr).
PS2
No de novo observation reported for NM_000257.3:c.2548G>A in any reviewed publication or ClinVar submission.
PS3
No variant-specific functional experimental data identified for c.2548G>A (p.Ala850Thr) in any reviewed publication.
PS4
No case-control data available with variant-specific counts for c.2548G>A.
PM5
A different missense variant at the same codon (p.Ala850Asp) is reported as a novel mutation in PMID:18409188, absent in 192 controls.
PM6
No assumed or confirmed de novo observation for c.2548G>A identified in any reviewed publication.
PP1
No segregation data available for this variant in any reviewed publication.
Benign
BA1
gnomAD v4.1 grpmax FAF = 7.9e-7, far below the VCEP BA1 threshold of ≥0.001.
BS1
gnomAD v4.1 grpmax FAF = 7.9e-7, far below the VCEP BS1 threshold of ≥0.0001 for MYH7.
BS3
No functional studies demonstrating a benign effect for c.2548G>A (p.Ala850Thr) identified in any reviewed publication.
BS4
No non-segregation data reported for this variant.
BP2
No evidence of this variant observed in cis or trans with a pathogenic variant in MYH7 or another cardiomyopathy gene.
BP4
REVEL score 0.809 is well above the VCEP BP4 threshold of ≤0.40.
N/A · 9
PVS1 · PP2 · PP4 · PP5 · BS2 · BP1 · BP5 · BP6 · BP7
Research & evidence
Population frequency
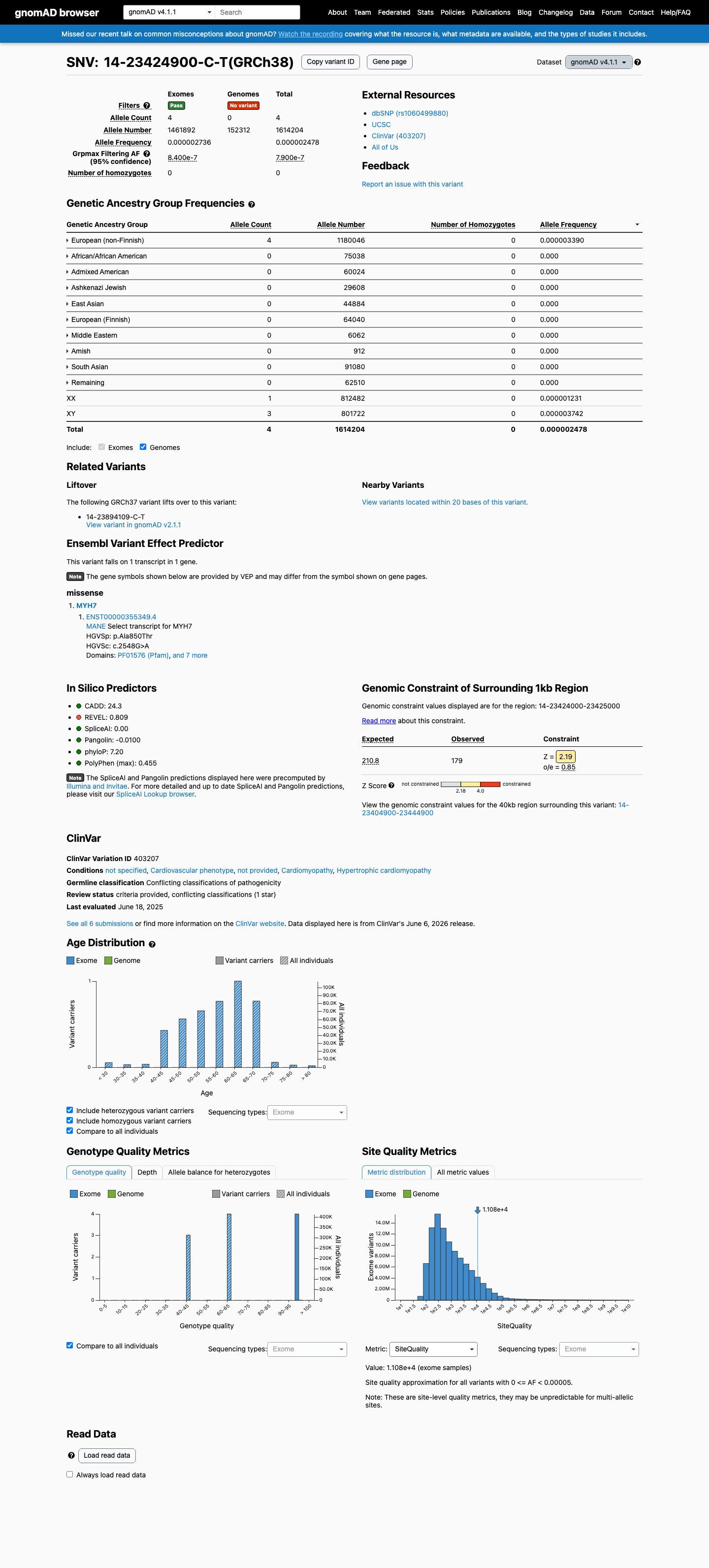
gnomAD v4.1
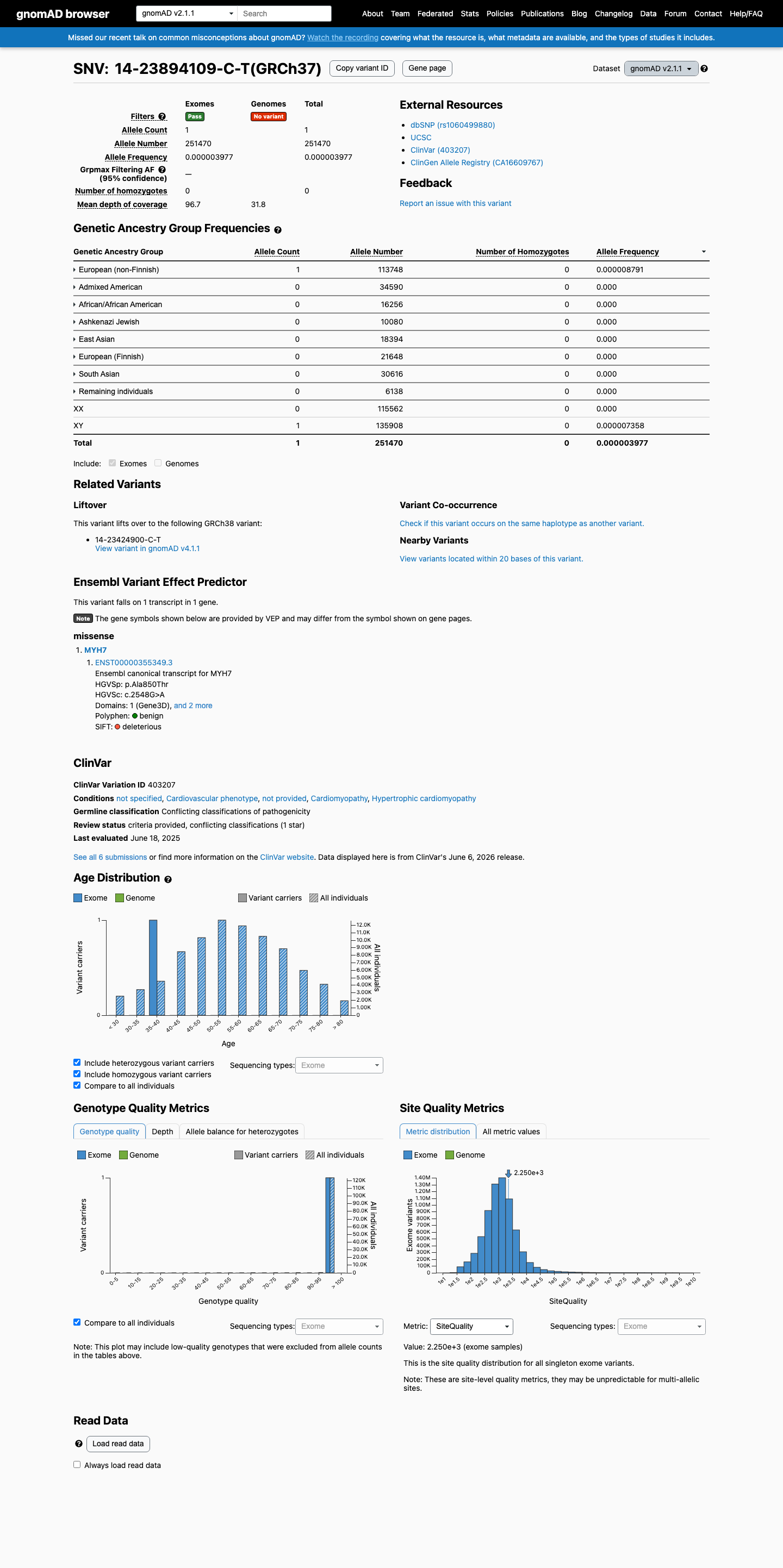
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.478e-06; MAF= 0.00025%, 4/1614204 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.3897e-06; MAF= 0.00034%, 4/1180046 alleles, homozygotes = 0); grpmax FAF= 7.9e-07.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.97662e-06; MAF= 0.00040%, 1/251470 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.79136e-06; MAF= 0.00088%, 1/113748 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,614,204
0 hom · FAF 7.9e-05%
0 hom · FAF 7.9e-05%
European (non-Finnish) 4 / 1,180,046 |
0.00034% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 251,470
0 hom
0 hom
European (non-Finnish) 1 / 113,748 |
0.00088% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
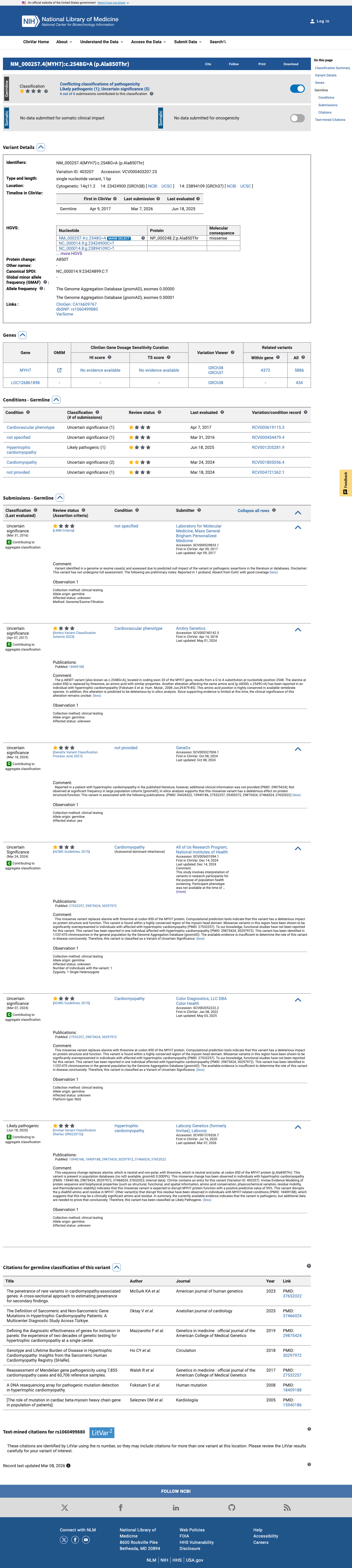
ClinVar
This variant has been reported in ClinVar as Uncertain significance (4 clinical laboratories) and as Uncertain Significance (1 clinical laboratory) and as Likely pathogenic (1 clinical laboratory). (ClinVarID = 403207)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.809. BayesDel score = 0.246045.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 9 PMIDs not cited in assessment
18409188 ↗
A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy.
CLINVAR
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
30297972 ↗
Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe).
CLINVAR
15940186 ↗
[The role of mutation in cardiac beta-myosin heavy chain gene in population of patients].
CLINVAR
27532257 ↗
Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples.
CLINVAR
29875424 ↗
Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center.
CLINVAR
37466024 ↗
The Definition of Sarcomeric and Non-Sarcomeric Gene Mutations in Hypertrophic Cardiomyopathy Patients: A Multicenter Diagnostic Study Across Türkiye.
CLINVAR
21810866 ↗
HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA).
CLINVAR