NM_000264.5:c.113G>A (p.Gly38Glu) is a missense variant in PTCH1. PVS1 is not applicable as the variant does not fall into a null-variant class.1 This variant is present in gnomAD population databases at frequencies (v2.1 AF=0.0096%, 25 alleles; v4.1 AF=0.0125%, 200 alleles) that exceed the maximum credible allele frequency for Gorlin syndrome (prevalence ~1/50,000-1/256,000), arguing against a highly penetrant pathogenic role.2 Multiple in silico predictors consistently suggest a benign effect: REVEL score 0.162, BayesDel score -0.434, and SpliceAI max delta 0.02 (BP4).3 The variant has been observed in a healthy adult carrier (mother of proband in Taeubner et al. 2018, PMID:29230040) who is heterozygous for PTCH1 p.G38E with no personal or family cancer history across three generations (BS2).4 The variant does not segregate with Gorlin syndrome features in the only reported family: the variant-carrying mother is unaffected, and the proband's congenital embryonal rhabdomyosarcoma occurred in the context of a co-occurring PTCH2 variant (BS4).5 Multiple clinical laboratories in ClinVar have classified this variant as Likely benign (4 labs) or Benign (1 lab), with only 2 reporting Uncertain significance and none reporting Pathogenic/Likely pathogenic (BP6).6 No pathogenic criteria are met. Applying the ACMG/AMP 2015 combination rules: 4 supporting benign criteria (BS2, BS4, BP4, BP6) are fulfilled, which classifies this variant as Likely benign.7
PTCH1
Final classification
Likely Benign
PTCH1 c.113G>A · p.Gly38Glu
PTCH1
NM_000264.5:c.113G>A (p.Gly38Glu) is a missense variant in PTCH1. PVS1 is not applicable as the variant does not fall into a null-variant class.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS2 supporting benign, BS4 supporting benign, BP4 supporting benign, BP6 supporting benign; combination = 4 supporting benign, which maps to Likely Benign.
Classification rationale
BS2BS4BP4BP6
Likely Benign
PTCH1 c.113G>A
BS2 + BS4 + BP4 + BP6
→
Likely Benign
1
pvs1_variant_assessment
3
revelbayesdelspliceai ↗
Gene diagram
· NM_000264.5 · variants mapped to exon structure
PTCH1
NM_000264.5
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 20 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
BS2
supporting
Benign
The variant has been observed in a healthy adult heterozygous carrier: the mother of the proband in PMID:29230040 carries the variant and has no cancer history, with an unremarkable family history spanning three generations. Additionally, the variant is present in gnomAD population databases (200 alleles in v4.1), indicating it is tolerated in the general population.
Healthy adult carrier (mother in PMID:29230040) heterozygous for PTCH1 p.G38E. No cancer historyunremarkable family history for 3 generations. Present in gnomAD v4.1 at 200 alleles.
✓
BS4
supporting
Benign
The variant does not segregate with disease in the only reported family (PMID:29230040). The variant was inherited from the mother, who has no personal or family history of Gorlin syndrome-associated features across three generations, while the proband presented with congenital embryonal rhabdomyosarcoma. This lack of segregation supports a benign interpretation.
Variant-carrier mother unaffectedunremarkable family history for 3 generations. Proband phenotype (congenital eRMS) not clearly attributable to PTCH1 p.G38E alone given co-occurring PTCH2 variant.
✓
BP4
supporting
Benign
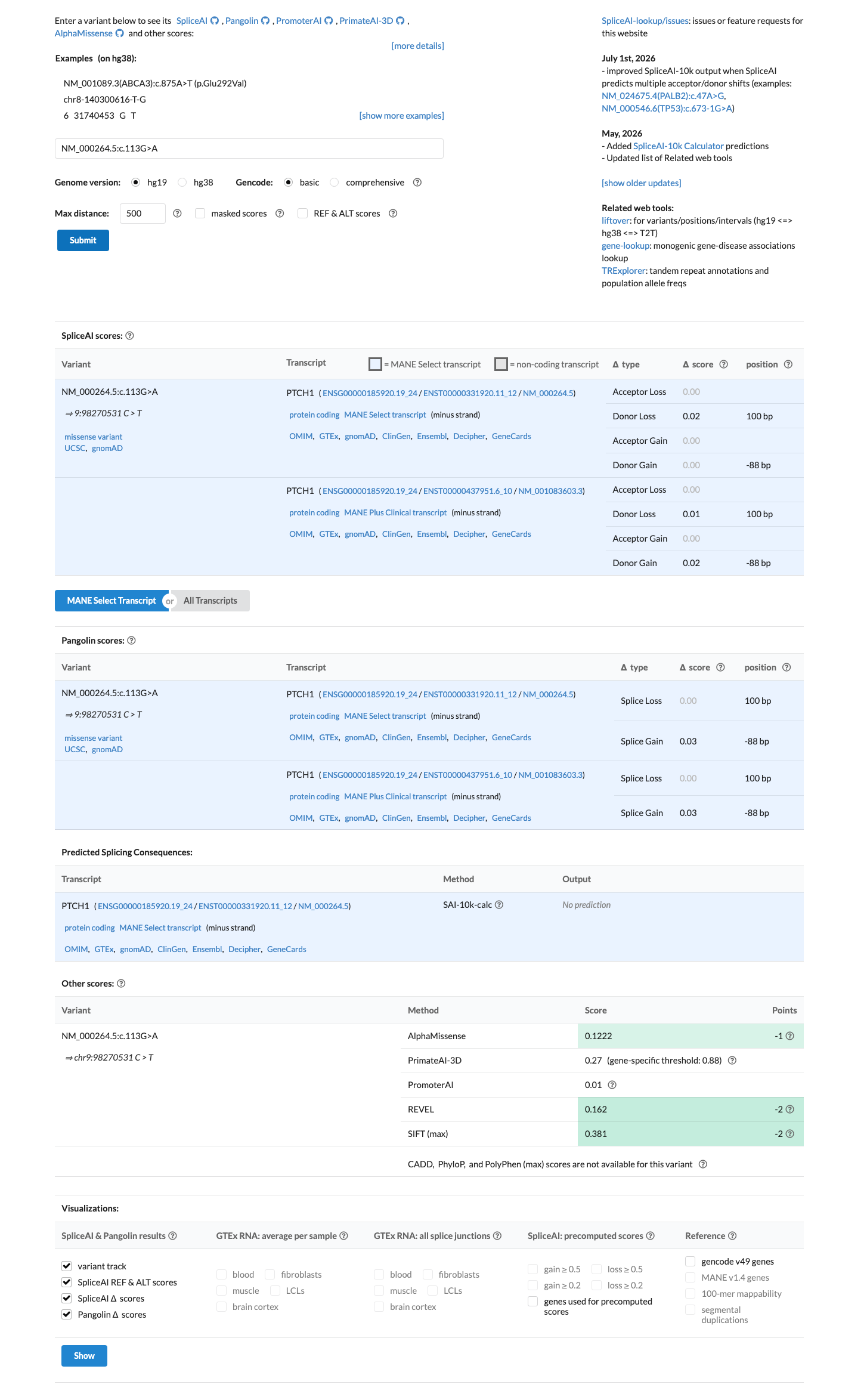
Multiple lines of computational evidence suggest no impact on gene product: REVEL score 0.162 (benign range, below 0.5 threshold), BayesDel score -0.434 (negative, predicting benign), and SpliceAI max delta score 0.02 (no predicted splicing impact).
REVEL=0.162 (benign)BayesDel=-0.434 (benign)SpliceAI max delta=0.02 (no splicing impact). Multiple concordant in silico predictions of benign effect.
✓
BP6
supporting
Benign
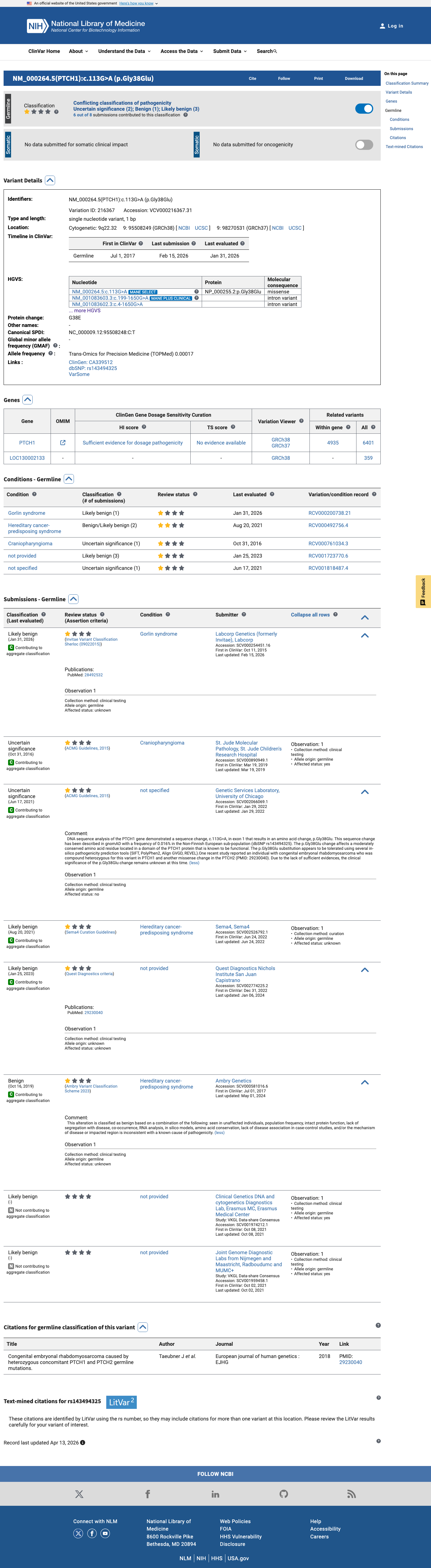
Multiple clinical laboratories have classified this variant as Likely benign or Benign in ClinVar (variation ID 216367): 4 labs report Likely benign, 1 reports Benign. Only 2 report Uncertain significance. No submitter reports Pathogenic or Likely pathogenic.
ClinVar variation 216367: Likely benign (4 labs)Benign (1 lab)VUS (2 labs). Majority clinical consensus supports benign interpretation.
Assessed · not applied
Pathogenic
PVS1
NM_000264.5:c.113G>A is a missense variant (p.Gly38Glu), not a null variant (nonsense, frameshift, or canonical splice site).
PS1
No alternate nucleotide change at the same amino acid position (p.Gly38) has been reported as pathogenic.
PS2
The variant was inherited from the mother, as confirmed by trio whole-exome sequencing in PMID:29230040.
PS3
No variant-specific functional assay demonstrating a deleterious effect has been performed.
PS4
The variant is present in gnomAD population databases at frequencies (v2.1: 0.0096%, v4.1: 0.0125%) that are inconsistent with a highly penetrant pathogenic variant for Gorlin syndrome (prevalence ~1/50,000-1/256,000).
PM1
p.Gly38 is located in the N-terminal extracellular region of PTCH1 (exon 1), N-terminal to the first transmembrane domain and well outside the sterol-sensing domain (SSD).
PM2
The variant is present in gnomAD population databases at frequencies inconsistent with absence from controls: v2.1 AF=0.0096% (25/259,610 alleles) and v4.1 AF=0.0125% (200/1,603,018 alleles).
PM5
No alternative pathogenic missense variant at the same amino acid residue (p.Gly38) was identified in ClinVar.
PM6
The variant is inherited from the mother, not de novo.
PP1
The variant does not cosegregate with disease in the only reported family (PMID:29230040).
PP2
No gene-level missense constraint data was available to establish a low rate of benign missense variation in PTCH1.
PP3
Multiple in silico predictors uniformly suggest a benign effect: REVEL score 0.162 (below 0.5 threshold), BayesDel score -0.434 (negative), and SpliceAI max delta 0.02 (no predicted splicing impact).
PP4
The proband in PMID:29230040 presented with congenital embryonal rhabdomyosarcoma, which can be part of the Gorlin syndrome spectrum.
PP5
No reputable source has classified this variant as pathogenic.
Benign
BA1
gnomAD allele frequency (v2.1: 0.0096%, v4.1: 0.0125%) is well below the BA1 threshold of >1%.
BS1
gnomAD allele frequency (v2.1: 0.0096%, v4.1: 0.0125%) is below the BS1 threshold of >0.3%.
BS3
No well-established in vitro or in vivo functional study has demonstrated that p.G38E has no damaging effect on protein function.
BP1
PTCH1 is a gene in which both missense and truncating variants are established mechanisms of disease for Gorlin syndrome.
BP2
No evidence that this variant has been observed in trans with a known pathogenic PTCH1 variant.
BP5
The proband in PMID:29230040 also harbored a PTCH2 p.His622Tyr variant, but this was described as a co-occurring/modifier variant, not an alternate molecular basis for the phenotype.
N/A · 1
BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
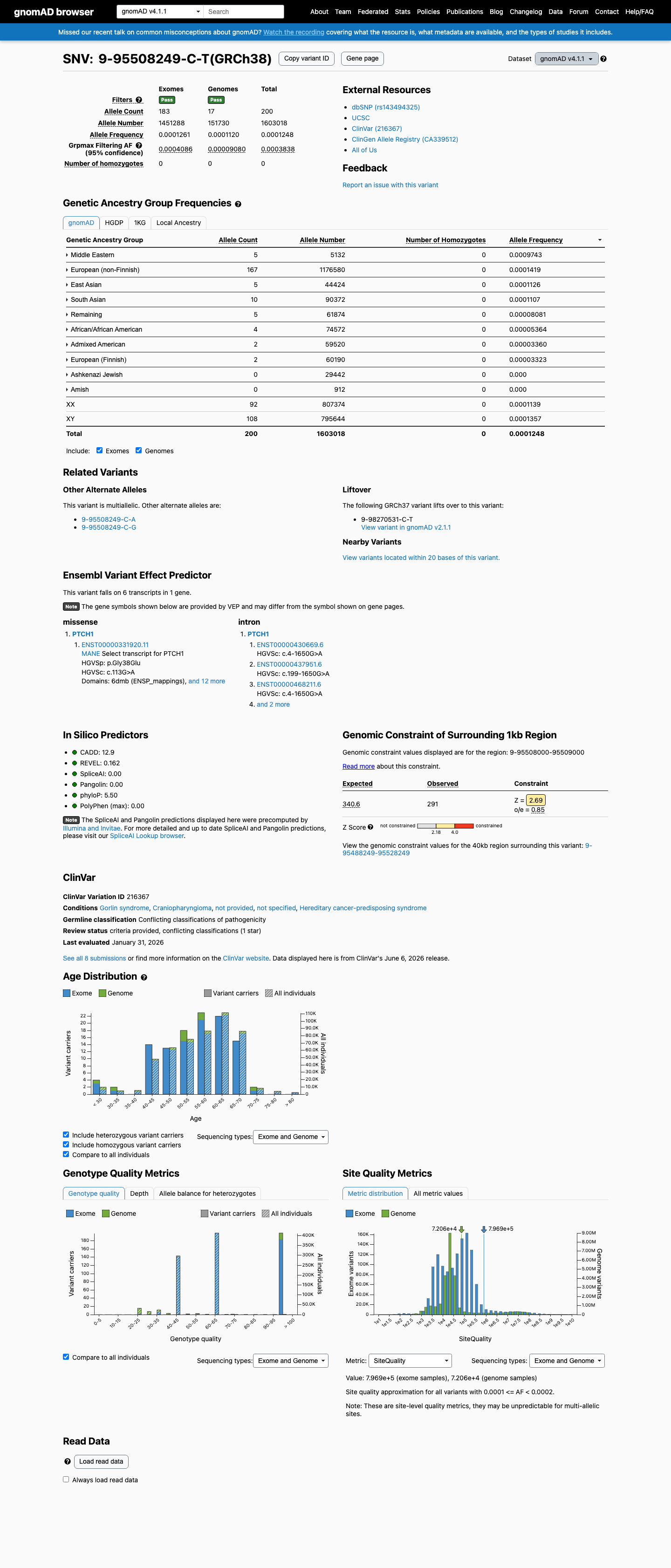
This variant is present in gnomAD v4.1 (AF= 0.000124765; MAF= 0.01248%, 200/1603018 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000974279; MAF= 0.09743%, 5/5132 alleles, homozygotes = 0); grpmax FAF= 0.00038378.
v2.1
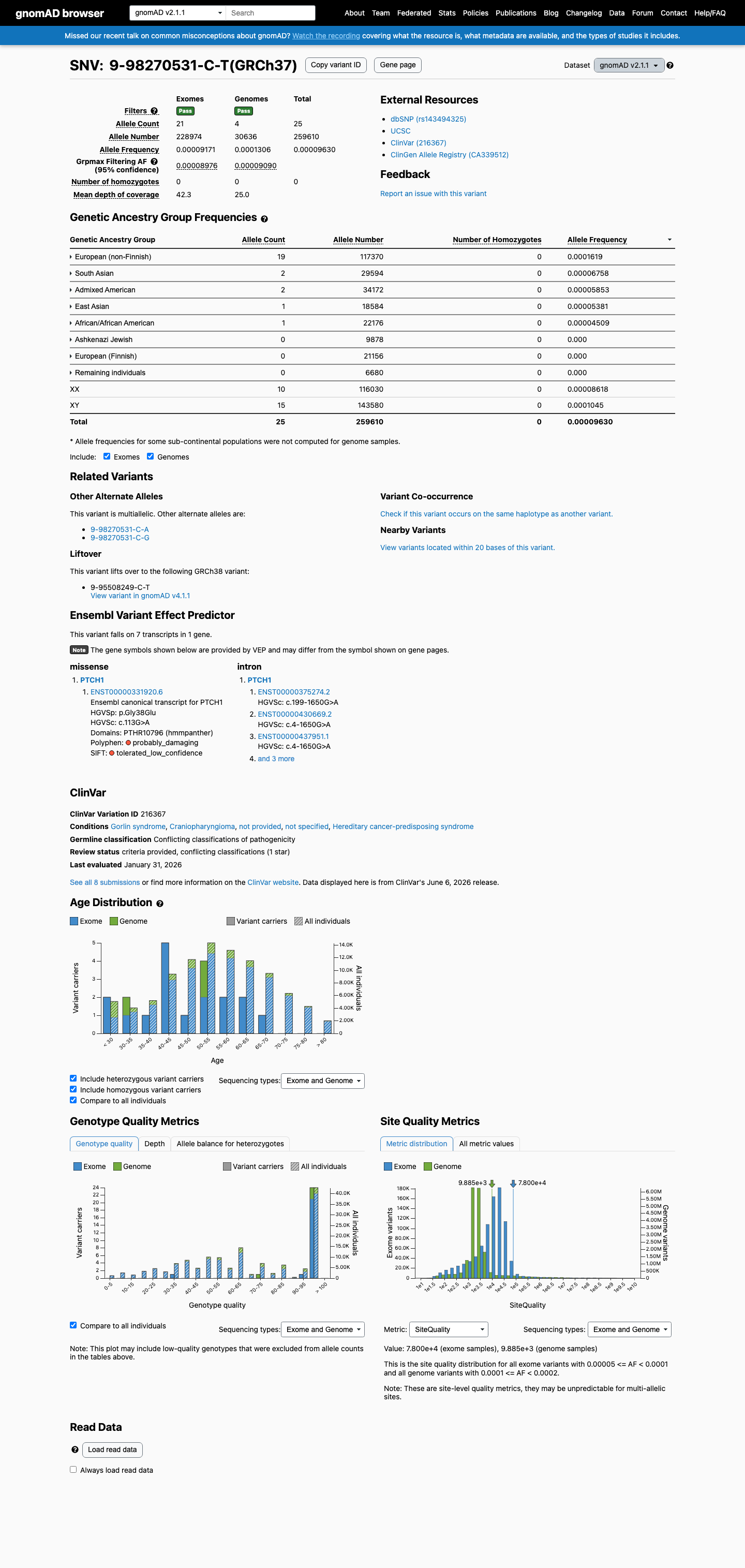
This variant is present in gnomAD v2.1 (AF= 9.62983e-05; MAF= 0.00963%, 25/259610 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.000161881; MAF= 0.01619%, 19/117370 alleles, homozygotes = 0); grpmax FAF= 9.09e-05.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0003266906239790918, 6/18366 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.012%
· 200 / 1,603,018
0 hom · FAF 0.038%
0 hom · FAF 0.038%
Middle Eastern 5 / 5,132 |
0.097% |
European (non-Finnish) 167 / 1,176,580 |
0.014% |
East Asian 5 / 44,424 |
0.011% |
South Asian 10 / 90,372 |
0.011% |
Remaining individuals 5 / 61,874 |
0.0081% |
African/African American 4 / 74,572 |
0.0054% |
Admixed American 2 / 59,520 |
0.0034% |
European (Finnish) 2 / 60,190 |
0.0033% |
+ 2 not observed (Amish, Ashkenazi Jewish)
gnomAD v2.1
0.0096%
· 25 / 259,610
0 hom · FAF 0.0091%
0 hom · FAF 0.0091%
European (non-Finnish) 19 / 117,370 |
0.016% |
South Asian 2 / 29,594 |
0.0068% |
Admixed American 2 / 34,172 |
0.0059% |
East Asian 1 / 18,584 |
0.0054% |
African/African American 1 / 22,176 |
0.0045% |
+ 3 not observed (Ashkenazi Jewish, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
0.033%
· 6 / 18,366
0 hom
0 hom
indel · split
Latino/Admixed American 1 / 834 |
0.12% |
European (non-Finnish) 5 / 11,712 |
0.043% |
+ 7 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
This variant has been reported in ClinVar as Likely benign (4 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 216367)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). REVEL score = 0.162. BayesDel score = -0.434182.
Functional
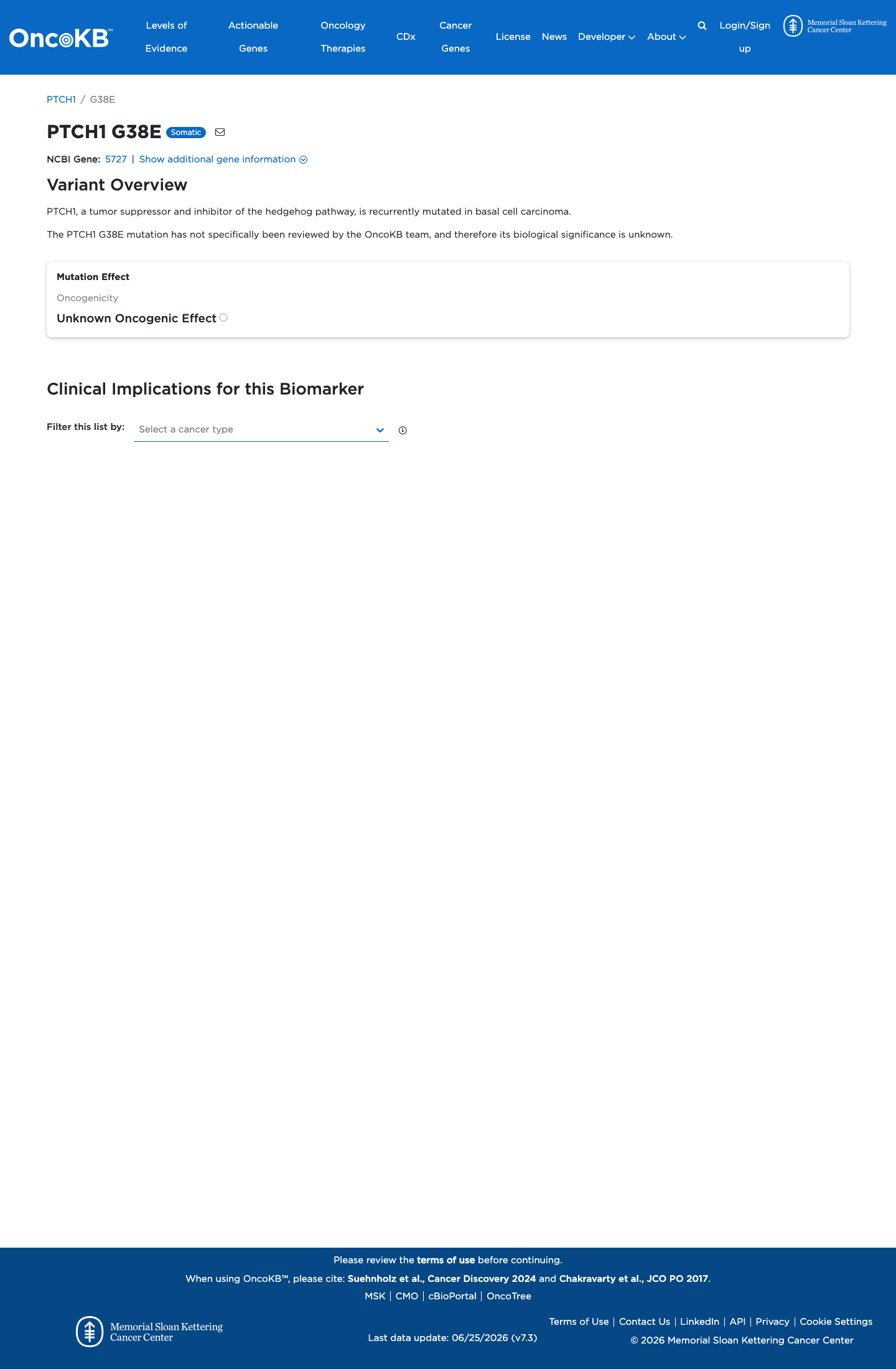
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. PTCH1, a tumor suppressor and inhibitor of the hedgehog pathway, is recurrently mutated in basal cell carcinoma.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
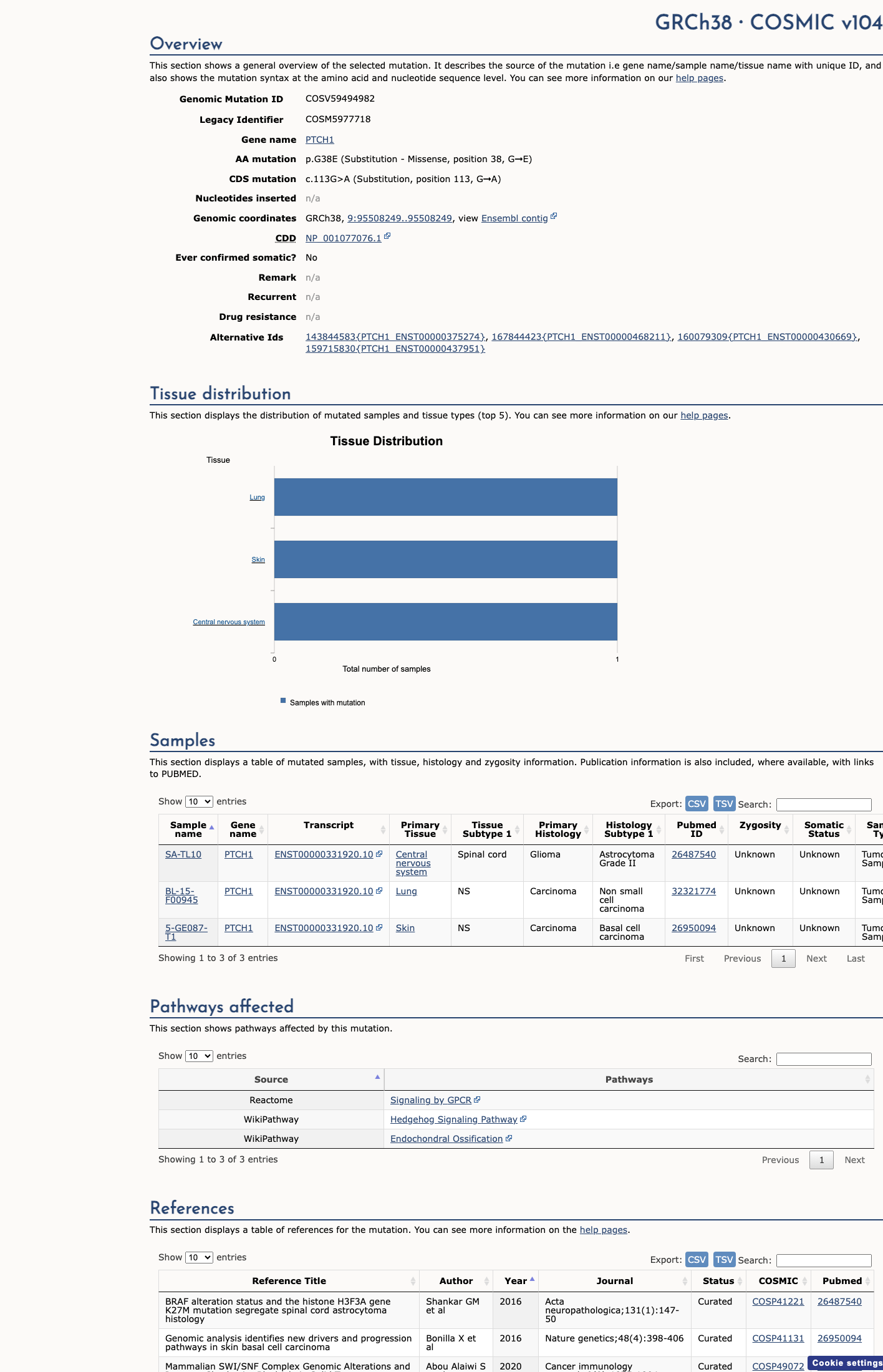
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV59494982, n = 3 times).
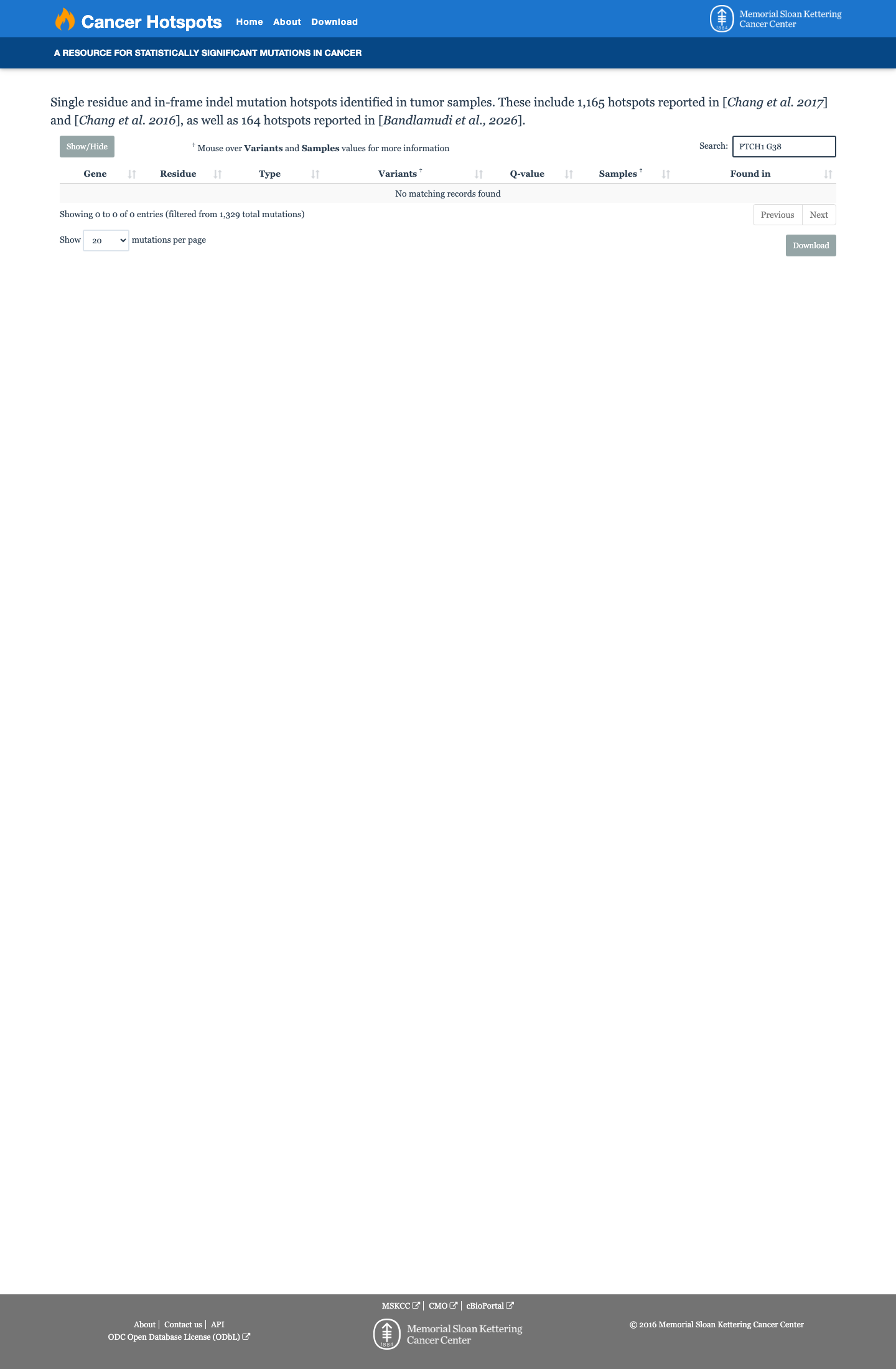
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
29230040 ↗
Congenital embryonal rhabdomyosarcoma caused by heterozygous concomitant PTCH1 and PTCH2 germline mutations.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
26389258 ↗
Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR