NM_000264.5:c.1641C>T is a synonymous variant (p.Ser547=) in PTCH1 with no predicted impact on splicing (SpliceAI max delta=0.00).1 This variant is present at high frequency in population databases: gnomAD v2.1 AF=0.798% (2242/281002 alleles, including 17 homozygotes) and v4.1 AF=1.125% (18153/1613520 alleles, including 125 homozygotes), exceeding the BA1 (>1%) and BS1 (>0.3%) thresholds for an autosomal dominant disorder with full penetrance expected at an early age.2 The observation of 17 homozygotes in gnomAD v2.1 and 125 homozygotes in gnomAD v4.1 is incompatible with a fully penetrant autosomal dominant condition; this alone is strong evidence for a benign classification (BS2).3 ClinVar classifies this variant as Benign based on submissions from 14 clinical laboratories; this represents a reputable source reporting a benign classification (BP6).4 The variant was identified as a non-pathogenic synonymous polymorphism (rs2066830, p.S547S) in a study of 78 sporadic medulloblastoma cases and in ovarian tumor/control cohorts, consistent with a benign interpretation.5 Computational evidence (SpliceAI) predicts no splicing impact; combined with its synonymous nature, this supports a benign interpretation (BP4, BP7).6 The BA1 criterion is met at stand-alone benign strength: allele frequency >1% in multiple gnomAD populations is incompatible with a highly penetrant dominant disorder. Per ACMG/AMP 2015 combination rules, meeting BA1 alone is sufficient for a Benign classification regardless of other criteria.7
PTCH1
Final classification
Benign
PTCH1 c.1641C>T · p.Ser547=
PTCH1
NM_000264.5:c.1641C>T is a synonymous variant (p.Ser547=) in PTCH1 with no predicted impact on splicing (SpliceAI max delta=0.00).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 strong benign, BP4 supporting benign, BP6 supporting benign, BP7 supporting benign; combination = 1 stand-alone benign + 2 strong benign + 3 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BS2BP4BP6BP7
Benign
PTCH1 c.1641C>T
BA1 + BS1 + BS2 + BP4 + BP6 + BP7
→
Benign
7
gnomad_v2 ↗gnomad_v4 ↗generic_acmg_combination_rules
Gene diagram
· NM_000264.5 · variants mapped to exon structure
PTCH1
NM_000264.5
Fetching transcript structure from UCSC…
Applied criteria · 6 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
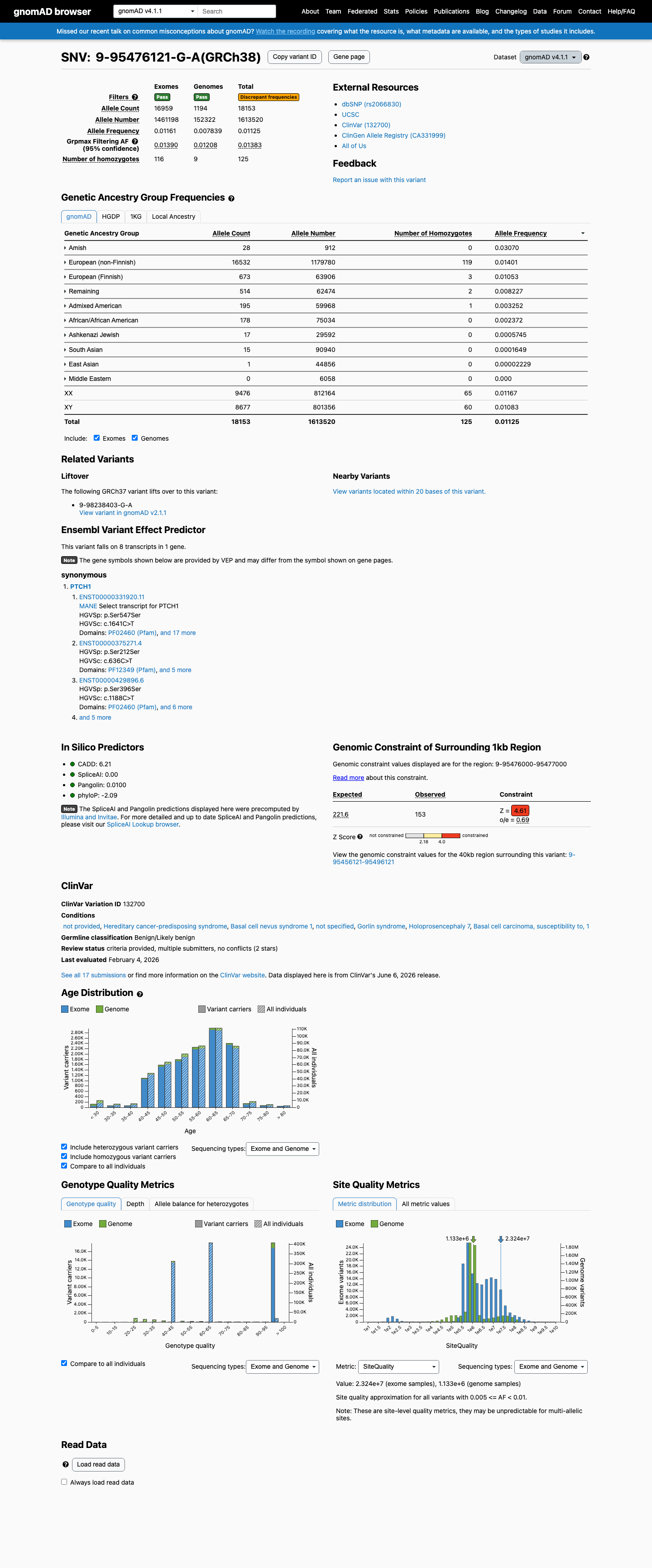
This variant is present in gnomAD v4.1 (AF= 0.0112506; MAF= 1.12506%, 18153/1613520 alleles, homozygotes = 125) and has highest observed frequency in the Amish population (AF= 0.0307018; MAF= 3.07018%, 28/912 alleles, homozygotes = 0); grpmax FAF= 0.0138338.
v2.1
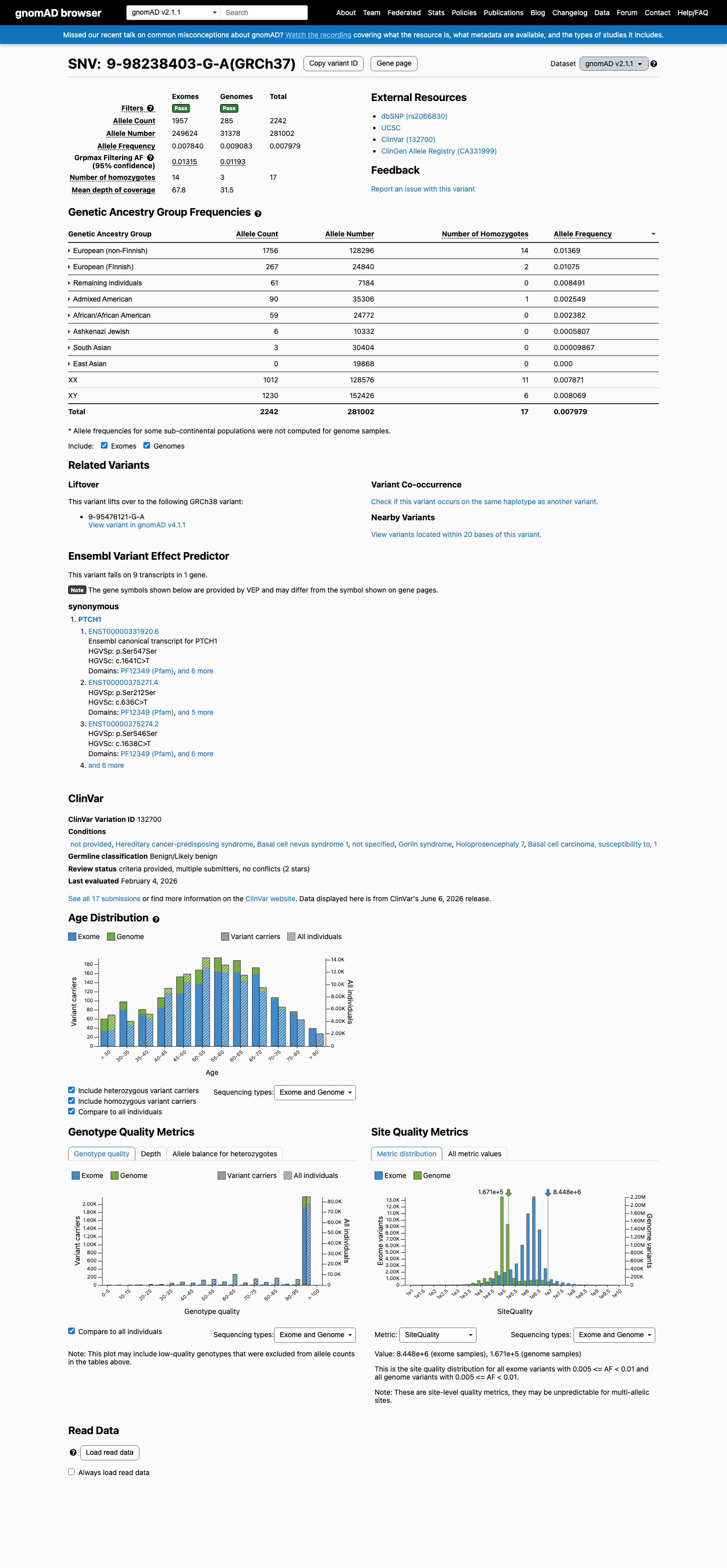
This variant is present in gnomAD v2.1 (AF= 0.00797859; MAF= 0.79786%, 2242/281002 alleles, homozygotes = 17) and has highest observed frequency in the European (non-Finnish) population (AF= 0.0136871; MAF= 1.36871%, 1756/128296 alleles, homozygotes = 14); grpmax FAF= 0.0131535.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.006785365324069048, 125/18422 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
1.1%
· 18153 / 1,613,520
125 hom · FAF 1.4%
125 hom · FAF 1.4%
Amish 28 / 912 |
3.1% |
European (non-Finnish) 16532 / 1,179,780 |
1.4% 119 hom |
European (Finnish) 673 / 63,906 |
1.1% 3 hom |
Remaining individuals 514 / 62,474 |
0.82% 2 hom |
Admixed American 195 / 59,968 |
0.33% 1 hom |
African/African American 178 / 75,034 |
0.24% |
Ashkenazi Jewish 17 / 29,592 |
0.057% |
South Asian 15 / 90,940 |
0.016% |
East Asian 1 / 44,856 |
0.0022% |
+ 1 not observed (Middle Eastern)
gnomAD v2.1
0.8%
· 2242 / 281,002
17 hom · FAF 1.3%
17 hom · FAF 1.3%
European (non-Finnish) 1756 / 128,296 |
1.4% 14 hom |
European (Finnish) 267 / 24,840 |
1.1% 2 hom |
Remaining individuals 61 / 7,184 |
0.85% |
Admixed American 90 / 35,306 |
0.25% 1 hom |
African/African American 59 / 24,772 |
0.24% |
Ashkenazi Jewish 6 / 10,332 |
0.058% |
South Asian 3 / 30,404 |
0.0099% |
+ 1 not observed (East Asian)
gnomAD Canada 🇨🇦
0.68%
· 125 / 18,422
0 hom · FAF 0.83%
0 hom · FAF 0.83%
European (non-Finnish) 115 / 11,742 |
0.98% |
Remaining individuals 6 / 1,138 |
0.53% |
Latino/Admixed American 2 / 838 |
0.24% |
Ashkenazi Jewish 1 / 832 |
0.12% |
African/African American 1 / 1,020 |
0.098% |
+ 4 not observed (East Asian, European (Finnish), Middle Eastern, South Asian)
ClinVar
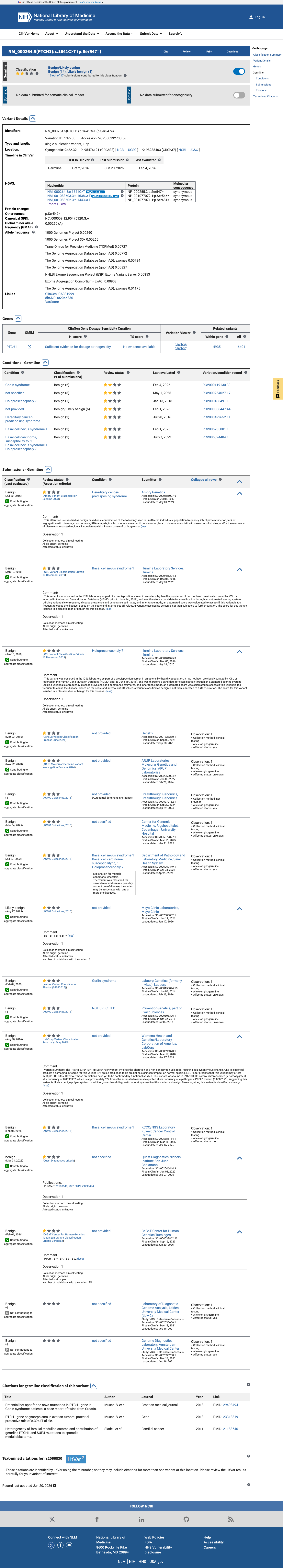
This variant has been reported in ClinVar as Benign (14 clinical laboratories) and as Likely benign (1 clinical laboratory) and as benign (1 clinical laboratory). (ClinVarID = 132700)
In silico
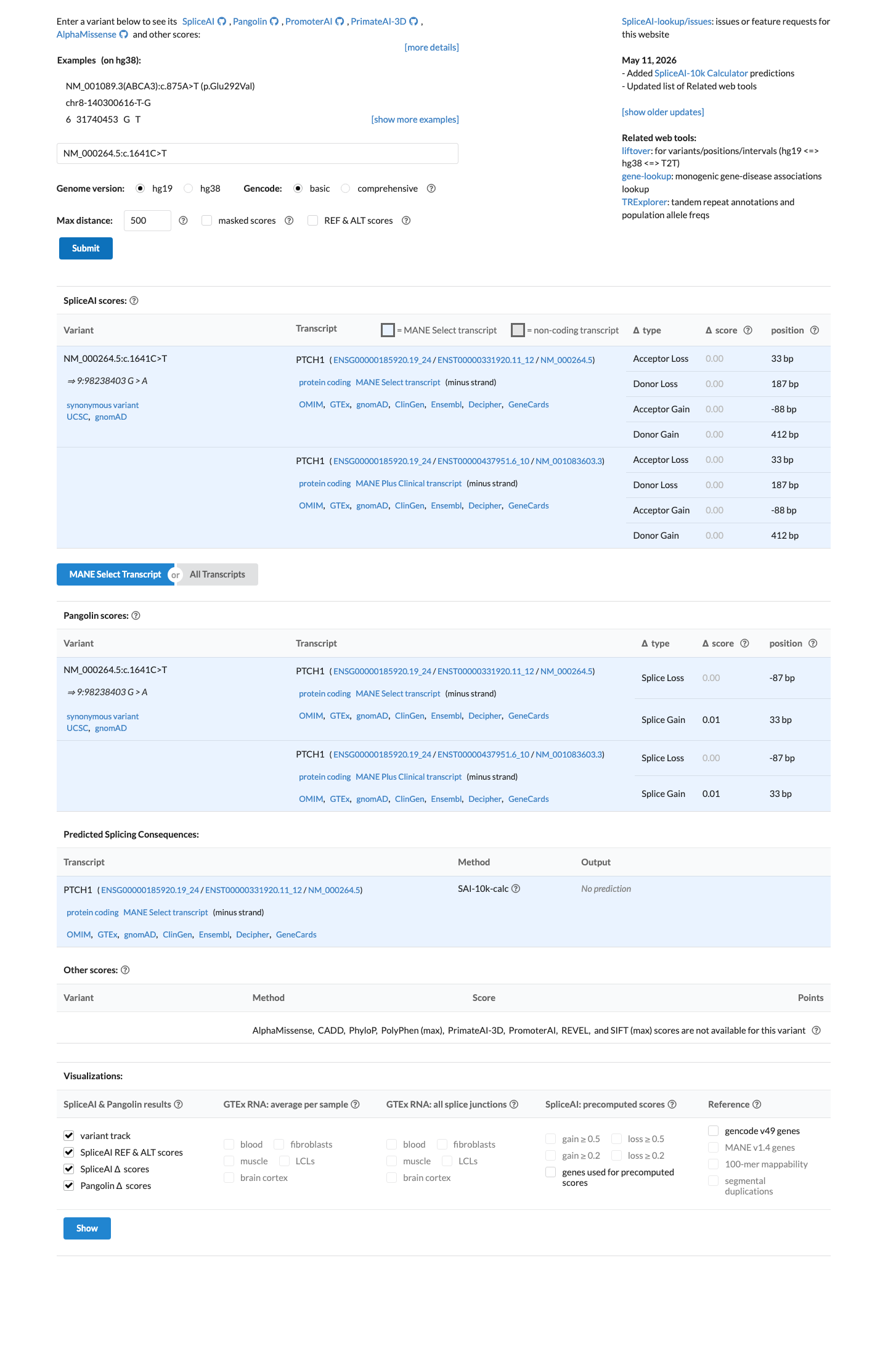
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV59461249, n = 5 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
21188540 ↗
Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
29498494 ↗
Potential hot spot for de novo mutations in PTCH1 gene in Gorlin syndrome patients: a case report of twins from Croatia.
CLINVAR
23313819 ↗
PTCH1 gene polymorphisms in ovarian tumors: potential protective role of c.3944T allele.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR