NM_000264.5:c.2438C>T (p.Pro813Leu) is a missense variant in exon 15 of PTCH1, a gene in which loss-of-function variants are an established cause of Gorlin syndrome (nevoid basal cell carcinoma syndrome).1 This variant is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (4/1,612,430 alleles, AF=2.48e-6, 0 homozygotes), meeting PM2 at moderate strength.2 Multiple lines of computational evidence suggest no significant impact on the gene product: BayesDel score is 0.0037 (strongly predicts benign) and SpliceAI max delta is 0.01 (no splicing impact), meeting BP4 at supporting benign level.3 The variant has been reported in ClinVar as Uncertain significance by a single clinical laboratory (Labcorp/Invitae, SCV005813886). It has been observed somatically in COSMIC (COSV59468245, n=2) but this does not inform germline pathogenicity.4 No variant-specific functional studies, segregation data, case-control analyses, or de novo observations were identified. Five literature-triage papers (PMIDs 15604628, 20301330, 21304560, 26389210, 26389333) were reviewed; none mention NM_000264.5:c.2438C>T.5 Applying generic ACMG/AMP 2015 combination rules (PMID:25741868): one moderate pathogenic criterion (PM2) and one supporting benign criterion (BP4) are present. These do not meet the threshold for Likely Pathogenic, Likely Benign, Pathogenic, or Benign. The variant is classified as a Variant of Uncertain Significance.6
PTCH1
Final classification
VUS
PTCH1 c.2438C>T · p.Pro813Leu
PTCH1
NM_000264.5:c.2438C>T (p.Pro813Leu) is a missense variant in exon 15 of PTCH1, a gene in which loss-of-function variants are an established cause of Gorlin syndrome (nevoid basal cell carcinoma syndrome).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 moderate, BP4 supporting benign; combination = 1 moderate + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
PTCH1 c.2438C>T
PM2 + BP4
→
VUS
Gene diagram
· NM_000264.5 · variants mapped to exon structure
PTCH1
NM_000264.5
Fetching transcript structure from UCSC…
Applied criteria · 2 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
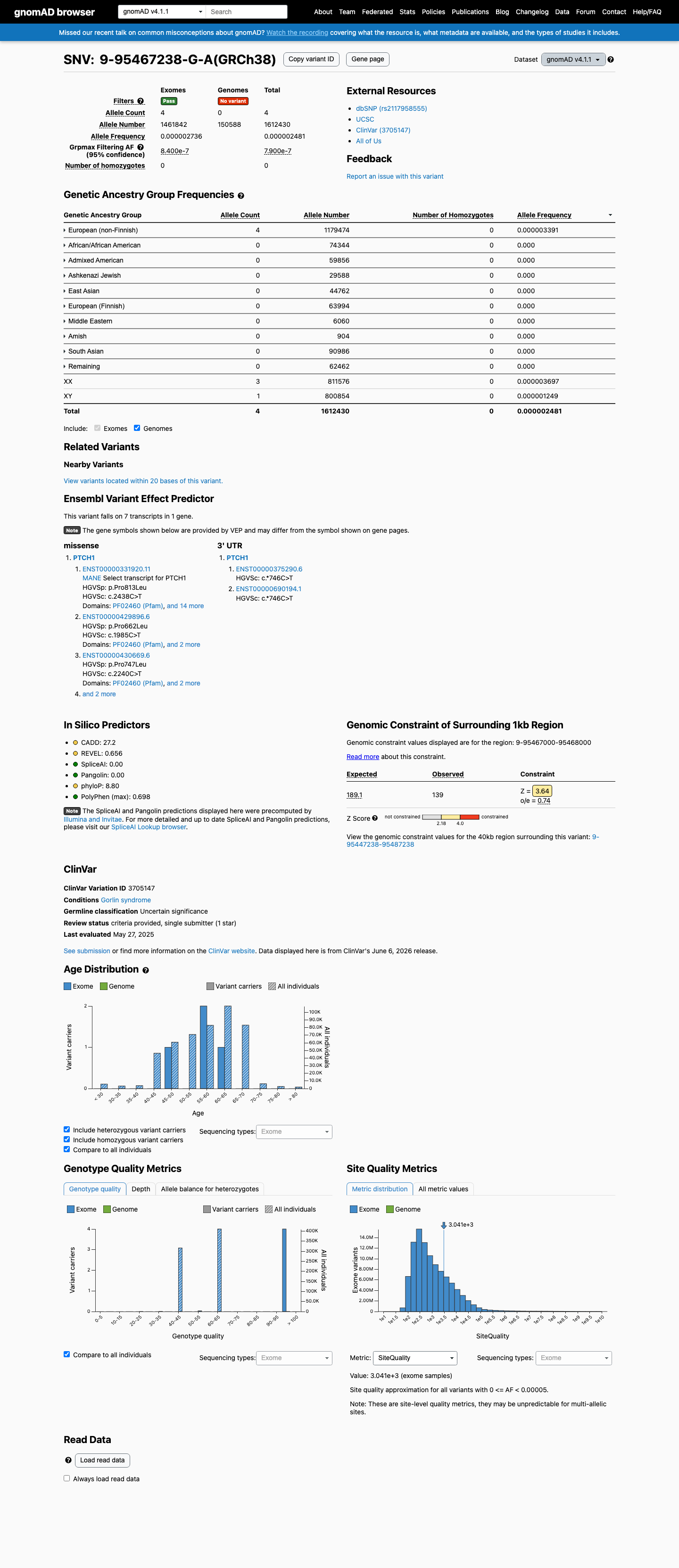
This variant is present in gnomAD v4.1 (AF= 2.48073e-06; MAF= 0.00025%, 4/1612430 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.39134e-06; MAF= 0.00034%, 4/1179474 alleles, homozygotes = 0); grpmax FAF= 7.9e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,612,430
0 hom · FAF 7.9e-05%
0 hom · FAF 7.9e-05%
European (non-Finnish) 4 / 1,179,474 |
0.00034% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
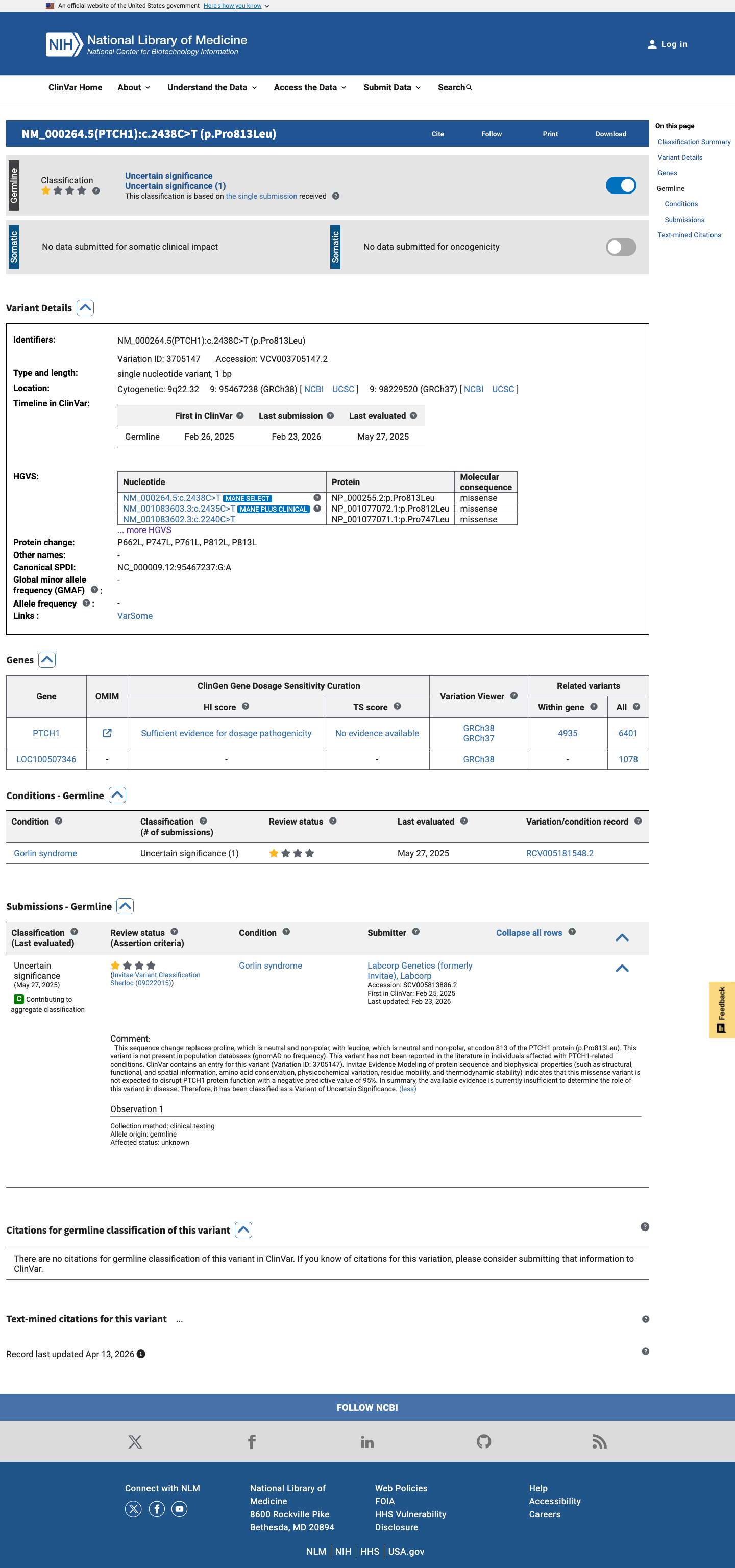
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory). (ClinVarID = 3705147)
In silico
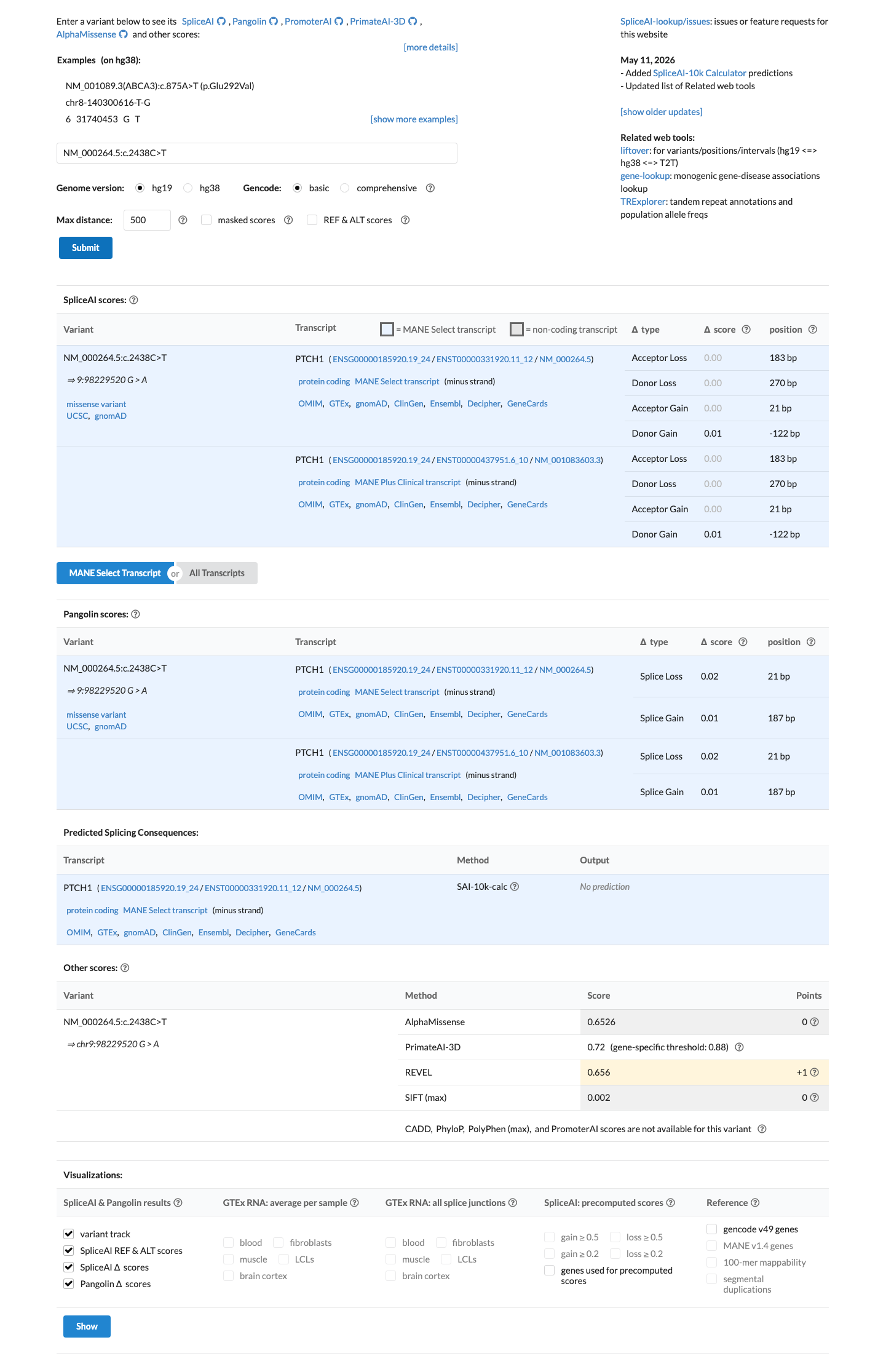
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.656. BayesDel score = 0.00372277.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. PTCH1, a tumor suppressor and inhibitor of the hedgehog pathway, is recurrently mutated in basal cell carcinoma.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV59468245, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR