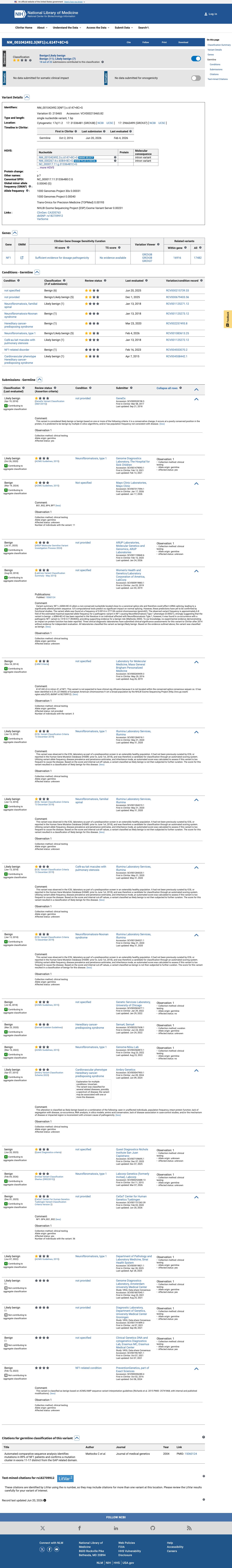
NM_000267.3:c.6084+8C>G is classified as Benign.1 BA1 is met at stand-alone benign strength: the variant has an allele frequency of 1.43% in the Amish subpopulation of gnomAD v4.1, exceeding the >1% BA1 threshold for a highly penetrant autosomal dominant disorder (NF1 prevalence ~1/3,000). Thirteen homozygous individuals are observed in gnomAD v4.1, which is incompatible with a pathogenic NF1 variant.2 BS1 is met at strong benign strength: the variant allele frequency of 1.43% (Amish subpopulation) far exceeds the 0.3% threshold for a dominant disorder.3 BP4 is met at supporting benign strength: SpliceAI predicts no splice impact (max delta = 0.00), and the variant is located at the +8 intronic position, distant from the canonical splice donor site.4 BP6 is met at supporting benign strength: ClinVar reports this variant as Benign based on 22 independent clinical laboratory submissions (11 Benign, 9 Likely benign, 1 benign).5 BA1 is stand-alone benign. Per ACMG/AMP 2015 combination rules (Richards et al., PMID:25741868), a single BA1 criterion is sufficient for a Benign classification. The final classification is Benign.6
NF1
Final classification
Benign
NF1 c.6084+8C>G · p.?
NF1
NM_000267.3:c.6084+8C>G is classified as Benign.
Neurofibromatosis and Schwannomatosis Specification v1.0.0 lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong benign, BP4 supporting benign, BP6 supporting benign; combination = 1 stand-alone benign + 1 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BP4BP6
Benign
NF1 c.6084+8C>G
BA1 + BS1 + BP4 + BP6
→
Benign
1
generic_acmg_combination_rules
6
generic_acmg_combination_rulesPMID:25741868 ↗
Gene diagram
· NM_000267.3 · variants mapped to exon structure
NF1
NM_000267.3
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 13 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
The variant has an allele frequency of 1.425% (13/912 alleles) in the Amish subpopulation in gnomAD v4.1, exceeding the 1% BA1 threshold. Additionally, 13 homozygotes are observed in gnomAD v4.1, which is incompatible with a highly penetrant autosomal dominant disorder such as NF1.
gnomAD v4.1 Amish subpopulation AF=1.425% (13/912 alleles) exceeds BA1 threshold of >1%13 homozygous individuals observed in gnomAD v4.1 overall. NF1 is an autosomal dominant disorder with near-complete penetrancehomozygosity for a pathogenic variant would be incompatible with life or result in exceptionally severe phenotype not seen in population databases.
✓
BS1
strong
Benign
The variant is present at an allele frequency of 1.425% in the Amish subpopulation of gnomAD v4.1, far exceeding the 0.3% BS1 threshold for a dominant disorder with NF1 prevalence (~1/3,000). The overall gnomAD v4.1 frequency of 0.29% also approaches this threshold.
gnomAD v4.1 Amish subpopulation AF=1.425% exceeds BS1 threshold of >0.3%. gnomAD v2.1 NFE AF=0.27%. Overall v4.1 AF=0.29% with 13 homozygotes. Population frequency incompatible with NF1 pathogenicity.
✓
BP4
supporting
Benign
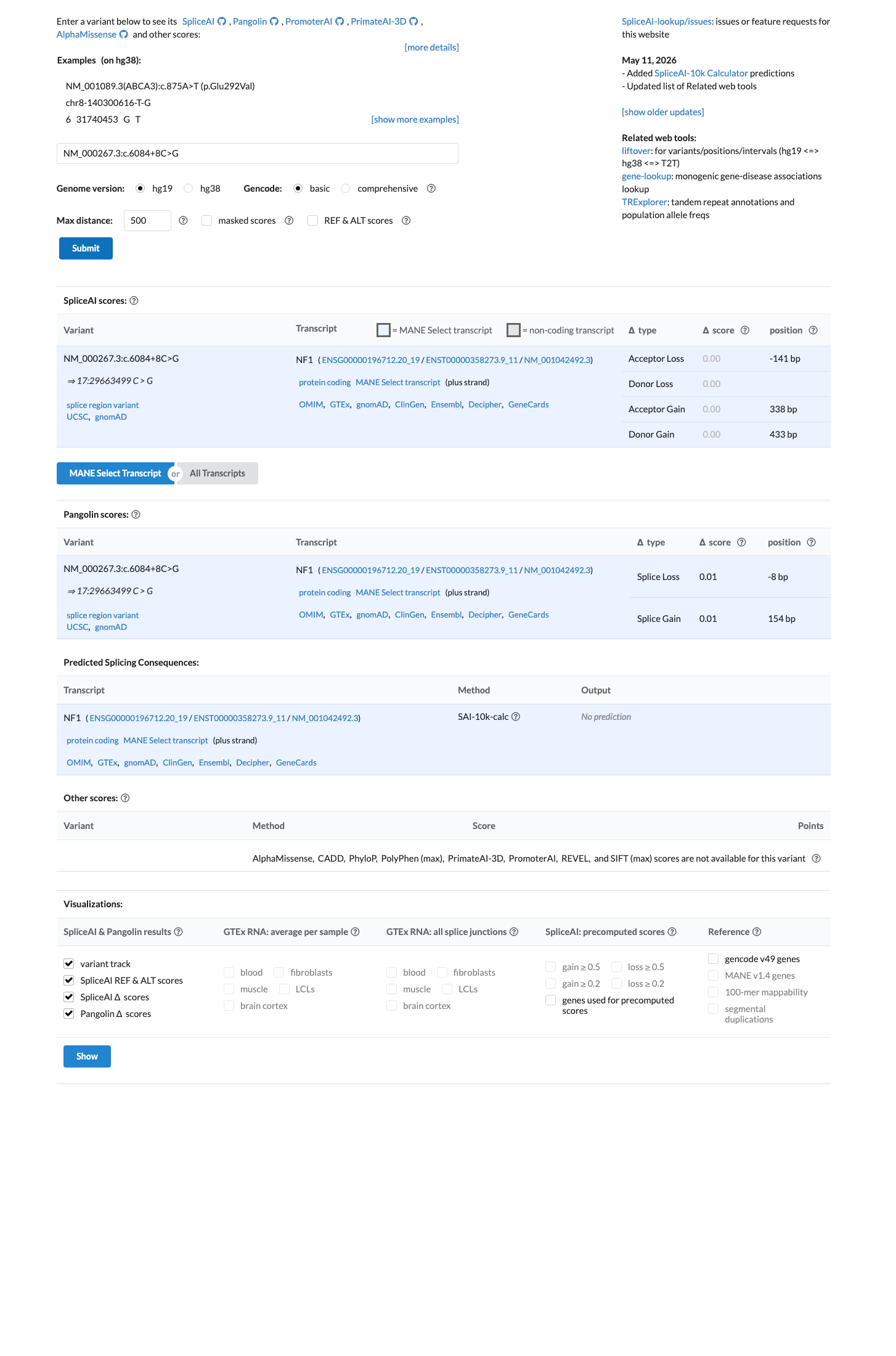
Multiple lines of computational evidence indicate no impact on splicing or gene function. SpliceAI predicts no splicing alteration (max delta score = 0.00). The variant is located deep within intron 40 at the +8 position, away from the canonical splice donor site.
SpliceAI max delta = 0.00 (no predicted donor lossdonor gainacceptor loss
✓
BP6
supporting
Benign
ClinVar reports this variant as Benign based on 22 submissions from multiple clinical laboratories (11 Benign, 9 Likely benign, 1 benign), though without expert panel review. Multiple independent clinical laboratories concur on a benign classification.
ClinVar Variation ID 215465: Benign22 submissions. Multiple independent clinical testing laboratories (GeneDxLabCorp
Assessed · not applied
Pathogenic
PS2
No confirmed de novo observation of NM_000267.3:c.6084+8C>G has been reported in any reviewed publication or ClinVar submission.
PS3
No well-established in vitro or in vivo functional studies demonstrating a damaging effect were identified.
PS4
The variant is present at high frequency in the general population (gnomAD v4.1: AF=0.29%, 4,673 alleles; Amish subpopulation AF=1.43%), precluding statistically significant case-control enrichment for a rare Mendelian disorder.
PM2
The variant is present in population databases at appreciable frequency (gnomAD v4.1: AF=0.29%, 4,673 alleles), far exceeding the PM2 threshold of <0.1%.
PP1
No segregation data are available for this variant in any reviewed publication or ClinVar submission.
PP3
Multiple lines of computational evidence support no splice impact.
PP4
No patient-specific phenotype data are available for this variant.
PP5
ClinVar reports this variant as Benign (22 submissions: 11 Benign, 9 Likely benign, 1 benign).
Benign
BS2
No confirmed healthy adult individuals with this variant have been reported with documented unaffected status.
BS3
No well-established functional studies demonstrating no damaging effect were identified.
BS4
No segregation data are available.
BP2
No observation of this variant in trans with a known pathogenic NF1 variant has been reported.
BP5
No case has been reported where this variant is found in an individual with an alternative molecular basis for NF1-related disease.
N/A · 9
PVS1 · PS1 · PM1 · PM5 · PM6 · PP2 · BP1 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
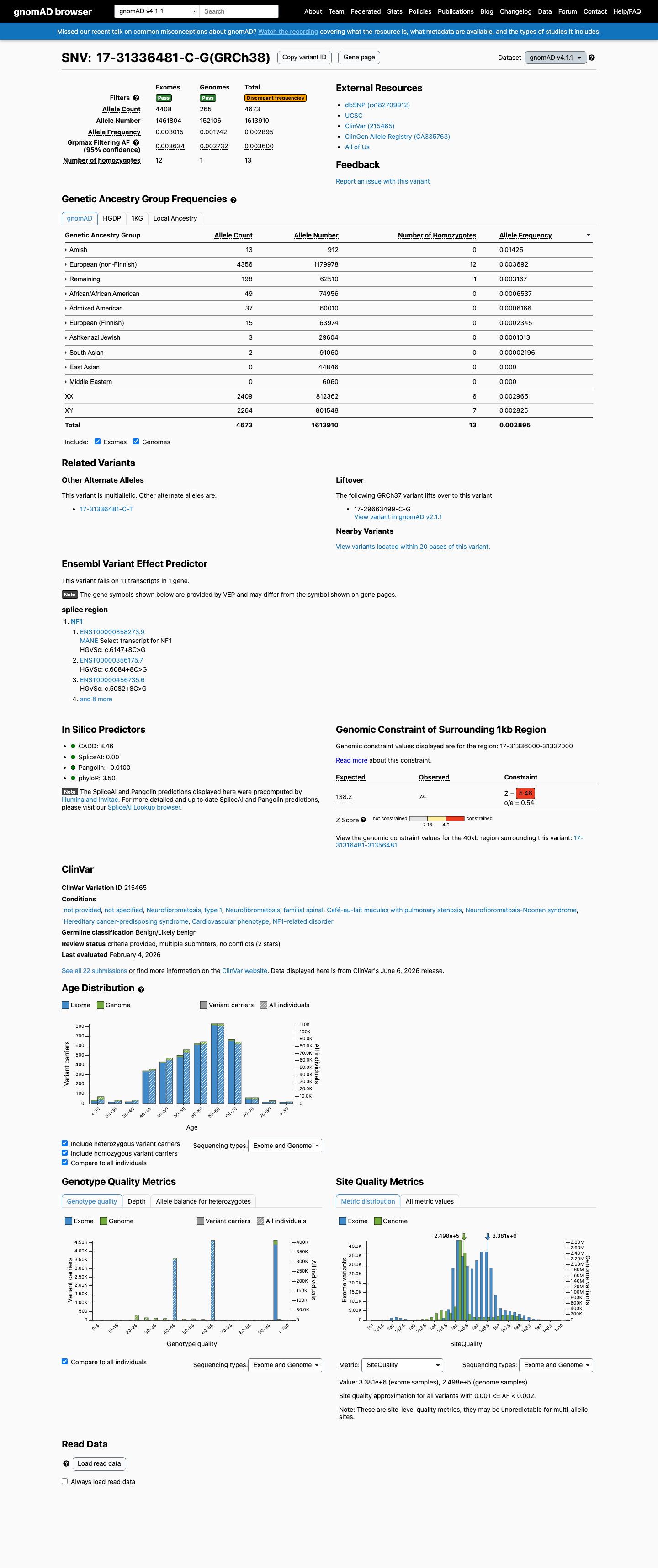
This variant is present in gnomAD v4.1 (AF= 0.00289545; MAF= 0.28955%, 4673/1613910 alleles, homozygotes = 13) and has highest observed frequency in the Amish population (AF= 0.0142544; MAF= 1.42544%, 13/912 alleles, homozygotes = 0); grpmax FAF= 0.00359997.
v2.1
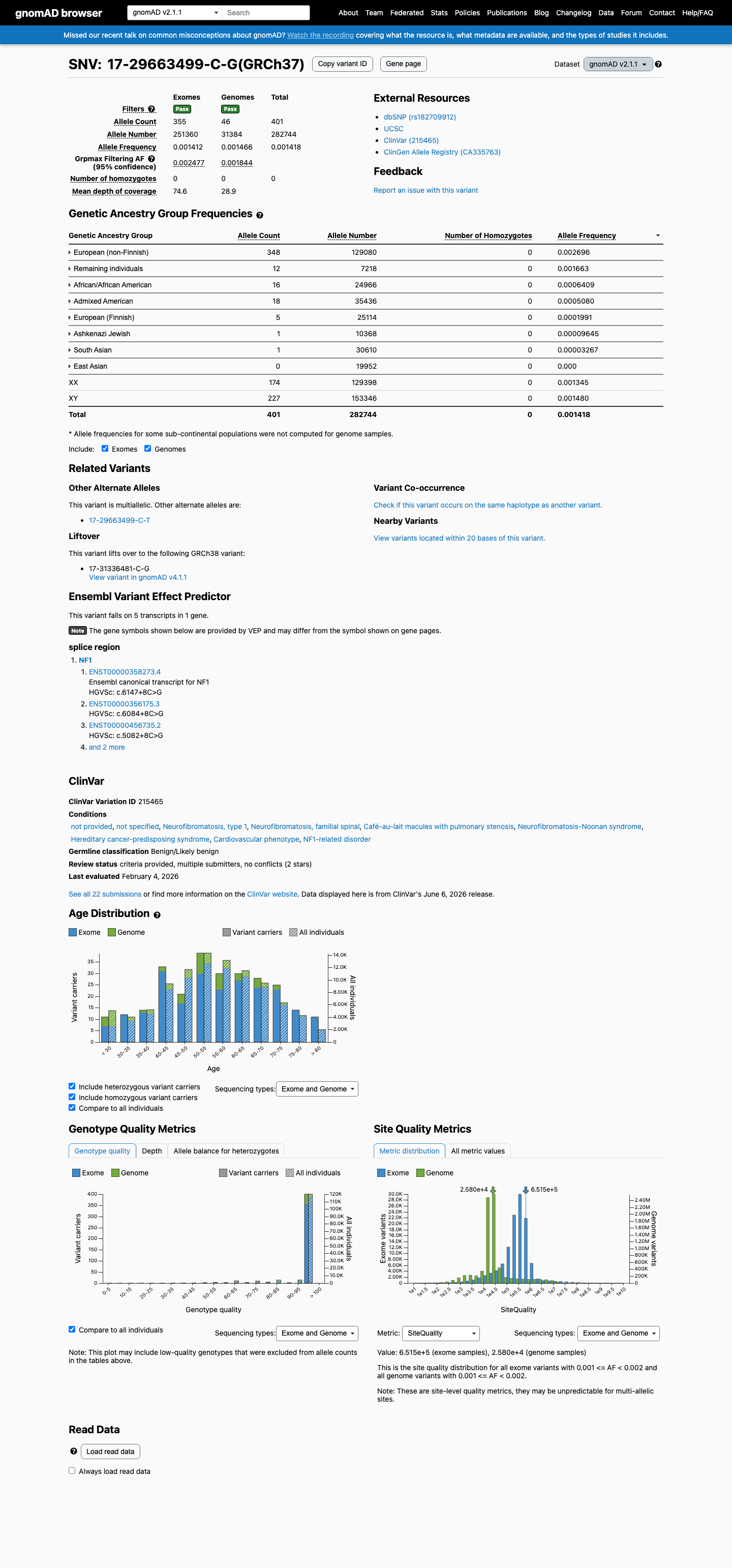
This variant is present in gnomAD v2.1 (AF= 0.00141824; MAF= 0.14182%, 401/282744 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 0.002696; MAF= 0.26960%, 348/129080 alleles, homozygotes = 0); grpmax FAF= 0.00247691.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.002500815483309775, 46/18394 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.29%
· 4673 / 1,613,910
13 hom · FAF 0.36%
13 hom · FAF 0.36%
Amish 13 / 912 |
1.4% |
European (non-Finnish) 4356 / 1,179,978 |
0.37% 12 hom |
Remaining individuals 198 / 62,510 |
0.32% 1 hom |
African/African American 49 / 74,956 |
0.065% |
Admixed American 37 / 60,010 |
0.062% |
European (Finnish) 15 / 63,974 |
0.023% |
Ashkenazi Jewish 3 / 29,604 |
0.01% |
South Asian 2 / 91,060 |
0.0022% |
+ 2 not observed (East Asian, Middle Eastern)
gnomAD v2.1
0.14%
· 401 / 282,744
0 hom · FAF 0.25%
0 hom · FAF 0.25%
European (non-Finnish) 348 / 129,080 |
0.27% |
Remaining individuals 12 / 7,218 |
0.17% |
African/African American 16 / 24,966 |
0.064% |
Admixed American 18 / 35,436 |
0.051% |
European (Finnish) 5 / 25,114 |
0.02% |
Ashkenazi Jewish 1 / 10,368 |
0.0096% |
South Asian 1 / 30,610 |
0.0033% |
+ 1 not observed (East Asian)
gnomAD Canada 🇨🇦
0.25%
· 46 / 18,394
0 hom · FAF 0.27%
0 hom · FAF 0.27%
European (non-Finnish) 41 / 11,718 |
0.35% |
Latino/Admixed American 2 / 836 |
0.24% |
Remaining individuals 2 / 1,138 |
0.18% |
Ashkenazi Jewish 1 / 832 |
0.12% |
+ 5 not observed (African/African American, East Asian, European (Finnish), Middle Eastern, South Asian)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 10 PMIDs not cited in assessment
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
15060124 ↗
Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11-17 distinct from the GAP related domain.
CLINVAR
20664475 ↗
The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer.
CLINVAR
26324357 ↗
American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility.
CLINVAR
24893135 ↗
Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline.
CLINVAR
26140447 ↗
Points to Consider: Ethical, Legal, and Psychosocial Implications of Genetic Testing in Children and Adolescents.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
33939658 ↗
The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma.
CLINVAR