BS1_Strong is met: the gnomAD v4.1 grpmax filtering allele frequency is 5.81e-05 (0.0058%), which falls within the PTEN VCEP BS1_Strong range of 0.0043%-0.056%. This population frequency is higher than expected for a fully penetrant pathogenic PTEN variant causing PHTS.1 BP7_Supporting is met: c.492+14dup is an intronic variant at position +14 (beyond +7). SpliceAI predicts no significant splice impact (max delta score 0.03) and no creation of a cryptic splice site, meeting the PTEN VCEP BP7 criteria.2 PM2_Supporting is not met: although the overall gnomAD allele frequency is very low (v4.1: 0.00025%), the Middle Eastern subpopulation in gnomAD v4.1 has 2 alleles out of 6,068 (AF 0.033%), which exceeds the PTEN VCEP subpopulation threshold of 0.002% for multiple alleles.3 PVS1 is not met: this intronic duplication at c.492+14 does not qualify as a null variant under the PTEN PVS1 decision tree, and SpliceAI predicts no splice disruption.4 Combined classification: 1 strong benign criterion (BS1) + 1 supporting benign criterion (BP7) yields a classification of Likely Benign per the ACMG/AMP combining rules adopted by the PTEN VCEP.5
PTEN
Final classification
Likely Benign
PTEN c.492+14dup · p.?
PTEN
BS1_Strong is met: the gnomAD v4.1 grpmax filtering allele frequency is 5.81e-05 (0.0058%), which falls within the PTEN VCEP BS1_Strong range of 0.0043%-0.056%. This population frequency is higher than expected for a fully penetrant pathogenic PTEN variant causing PHTS.
Richards et.al., 2015 - Combining rules v3.2.0 criteria-combination framework: matched Rule20 (1 Benign.Strong) with applied criteria: BS1 strong benign, BP7 supporting benign; maps to Likely Benign.
Classification rationale
BS1BP7
Likely Benign
PTEN c.492+14dup
BS1 + BP7
→
Likely Benign
Gene diagram
· NM_000314.8 · variants mapped to exon structure
PTEN
NM_000314.8
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 15 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
BS1_Strong under PTEN VCEP applies when gnomAD filtering allele frequency is 0.0043% to 0.056%. The gnomAD v4.1 grpmax filtering AF for c.492+14dup is 5.81e-05 (0.0058%), which falls within the BS1_Strong range (0.0043%-0.056%). This variant is observed at a population frequency higher than expected for a fully penetrant PTEN pathogenic variant causing PHTS.
gnomAD v4.1 grpmax FAF = 5.81e-05 (0.0058%)within PTEN VCEP BS1_Strong range of 0.0043%-0.056%.
✓
BP7
supporting
Benign
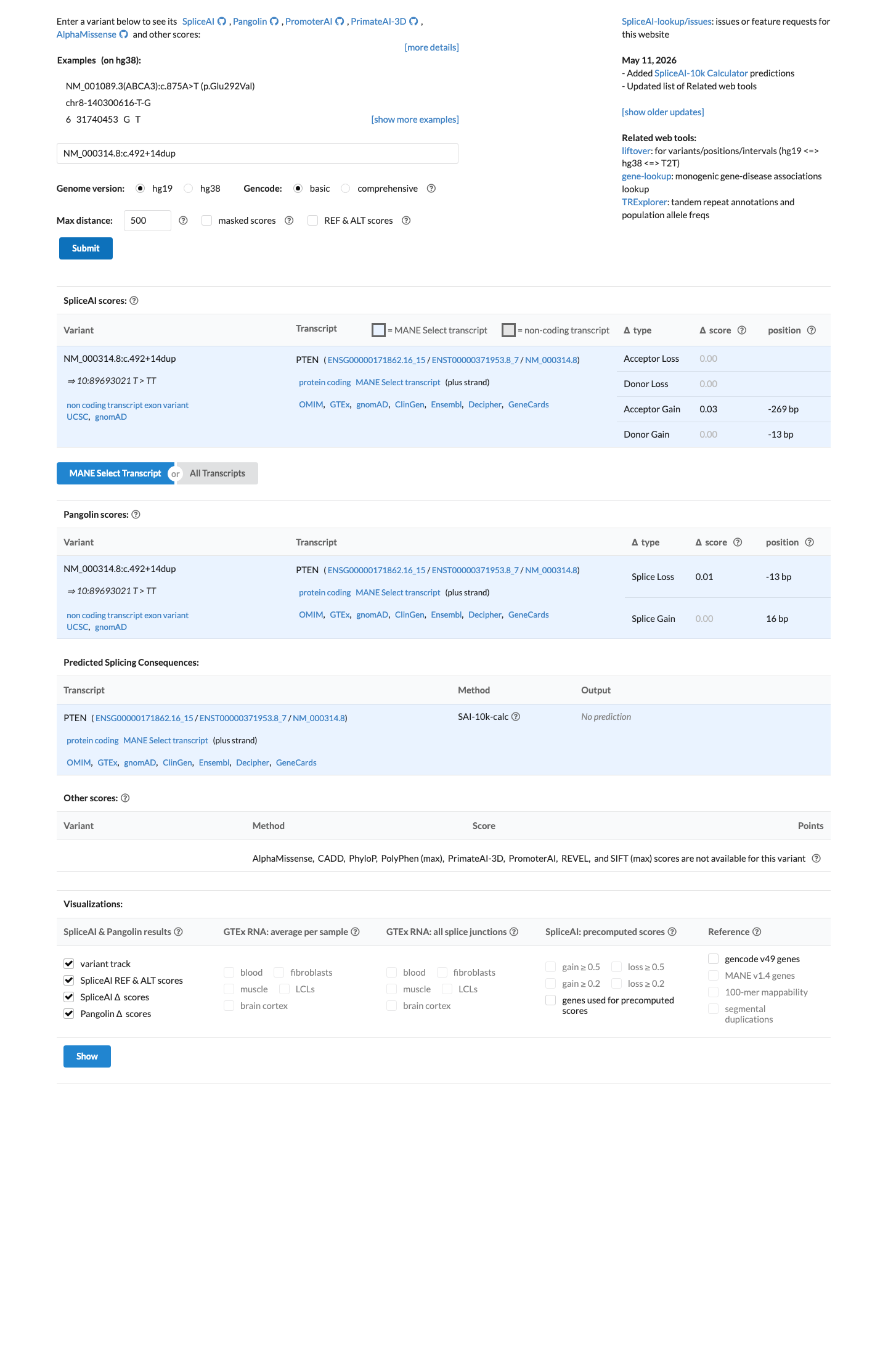
BP7 under PTEN VCEP applies to intronic variants at or beyond +7/-21 for which splicing prediction algorithms predict no impact. c.492+14dup is located at position +14 in intron 5 (beyond the +7 threshold). SpliceAI predicts no significant splice impact (max delta score 0.03), and the variant does not create a new splice site.
Intronic position +14 (beyond +7 threshold).SpliceAI max delta 0.03 — predicts no splice impact.No cryptic splice site creation predicted.
Assessed · not applied
Pathogenic
PVS1
PVS1 is not applicable to this intronic variant (c.492+14dup).
PS2
No de novo observations of NM_000314.8:c.492+14dup have been identified in ClinVar, LOVD, or published literature.
PS3
No RNA, mini-gene, or splicing functional assay data are available for c.492+14dup.
PS4
No proband counts, specificity scores, or case-control studies are available for this variant.
PM2
The overall gnomAD frequency is very low (v4.1: 4/1,601,512, AF = 0.00025%), meeting the PTEN VCEP main threshold of <0.001%.
PM6
No assumed de novo observations of c.492+14dup have been reported in ClinVar, LOVD, or published literature.
PP1
No co-segregation data are available for c.492+14dup.
PP3
PP3 under PTEN VCEP requires SpliceAI scores of 0.5-1.0 for splicing variants (with VarSeak concordance).
Benign
BA1
BA1 under PTEN VCEP requires gnomAD filtering allele frequency >0.056% (0.00056).
BS2
BS2 under PTEN VCEP requires observation of the variant in the homozygous state in a healthy or PHTS-unaffected individual.
BS3
BS3_Strong under PTEN VCEP for intronic variants requires RNA/mini-gene assay demonstrating no splicing impact.
BS4
No segregation data are available for c.492+14dup.
BP2
No observations of c.492+14dup in trans with a pathogenic/likely pathogenic PTEN variant or in cis with multiple P/LP PTEN variants have been reported.
BP4
BP4 under PTEN VCEP requires concordance of SpliceAI and VarSeak predicting no splicing impact (SpliceAI 0-0.2, VarSeak Class 1-2).
BP5
BP5 under PTEN VCEP requires at least two cases where the variant is found with an alternate molecular basis for disease, with no phenotypic overlap.
N/A · 10
PS1 · PM1 · PM4 · PM5 · PP2 · PP4 · PP5 · BP1 · BP3 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
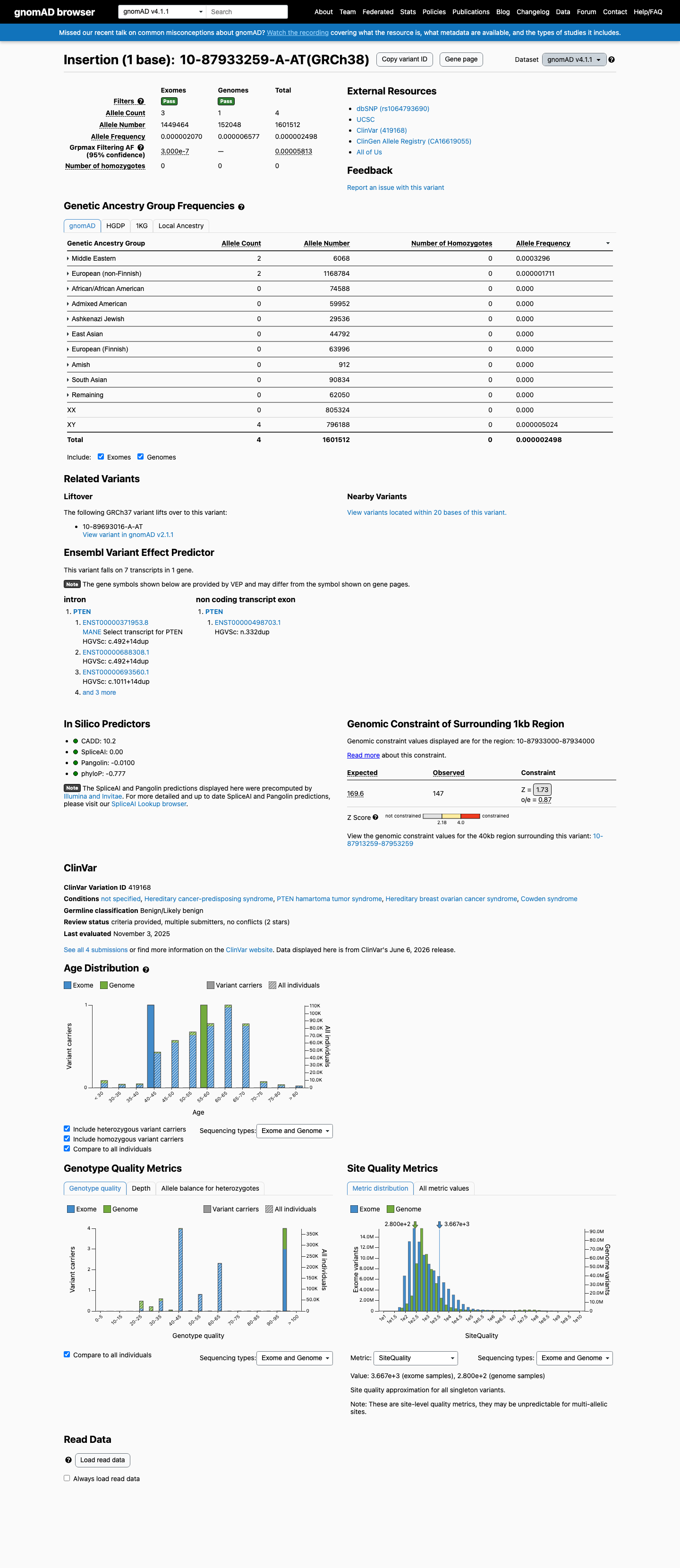
This variant is present in gnomAD v4.1 (AF= 2.49764e-06; MAF= 0.00025%, 4/1601512 alleles, homozygotes = 0) and has highest observed frequency in the Middle Eastern population (AF= 0.000329598; MAF= 0.03296%, 2/6068 alleles, homozygotes = 0); grpmax FAF= 5.813e-05.
v2.1
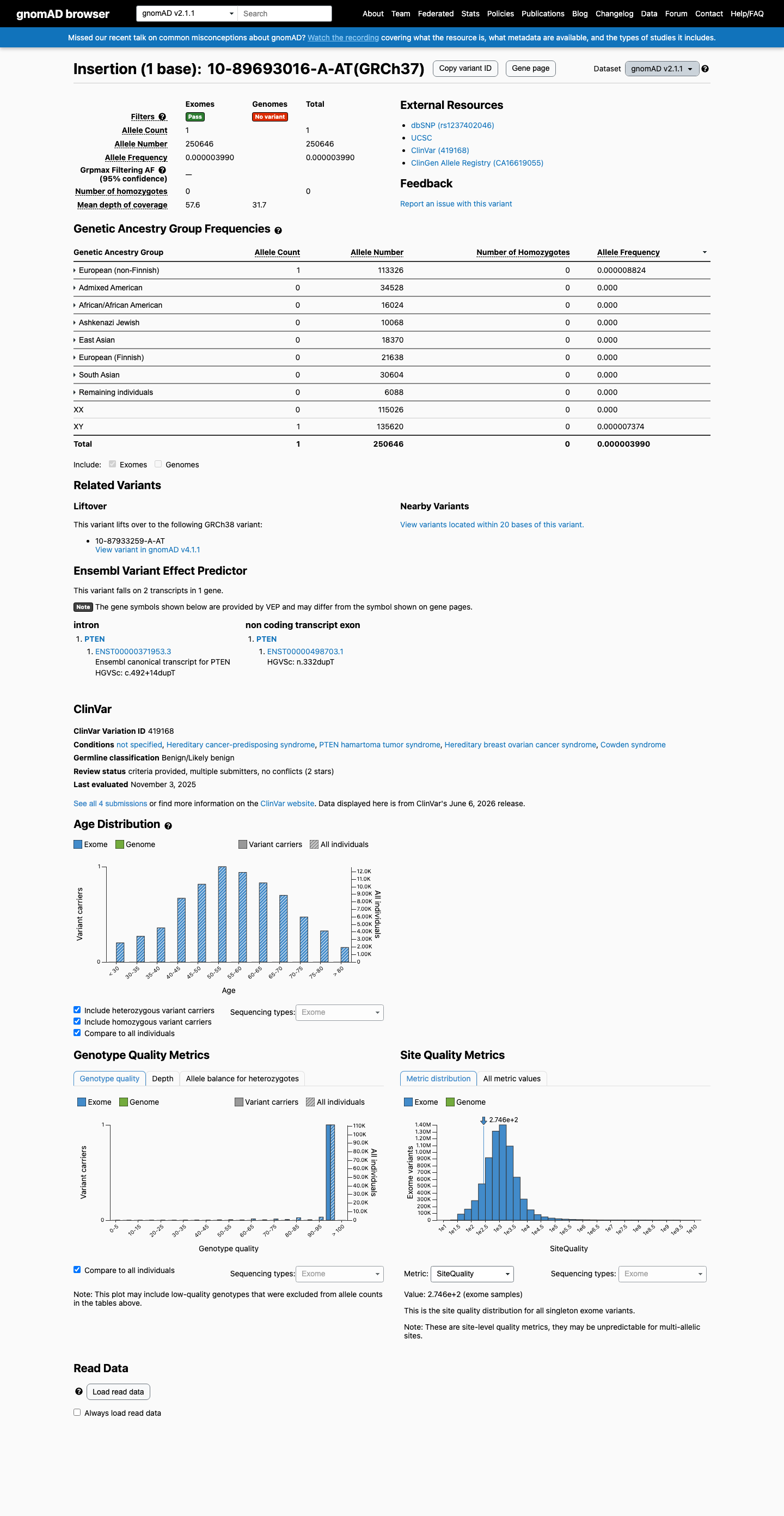
This variant is present in gnomAD v2.1 (AF= 3.98969e-06; MAF= 0.00040%, 1/250646 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.8241e-06; MAF= 0.00088%, 1/113326 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,601,512
0 hom · FAF 0.0058%
0 hom · FAF 0.0058%
Middle Eastern 2 / 6,068 |
0.033% |
European (non-Finnish) 2 / 1,168,784 |
0.00017% |
+ 8 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 250,646
0 hom
0 hom
European (non-Finnish) 1 / 113,326 |
0.00088% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
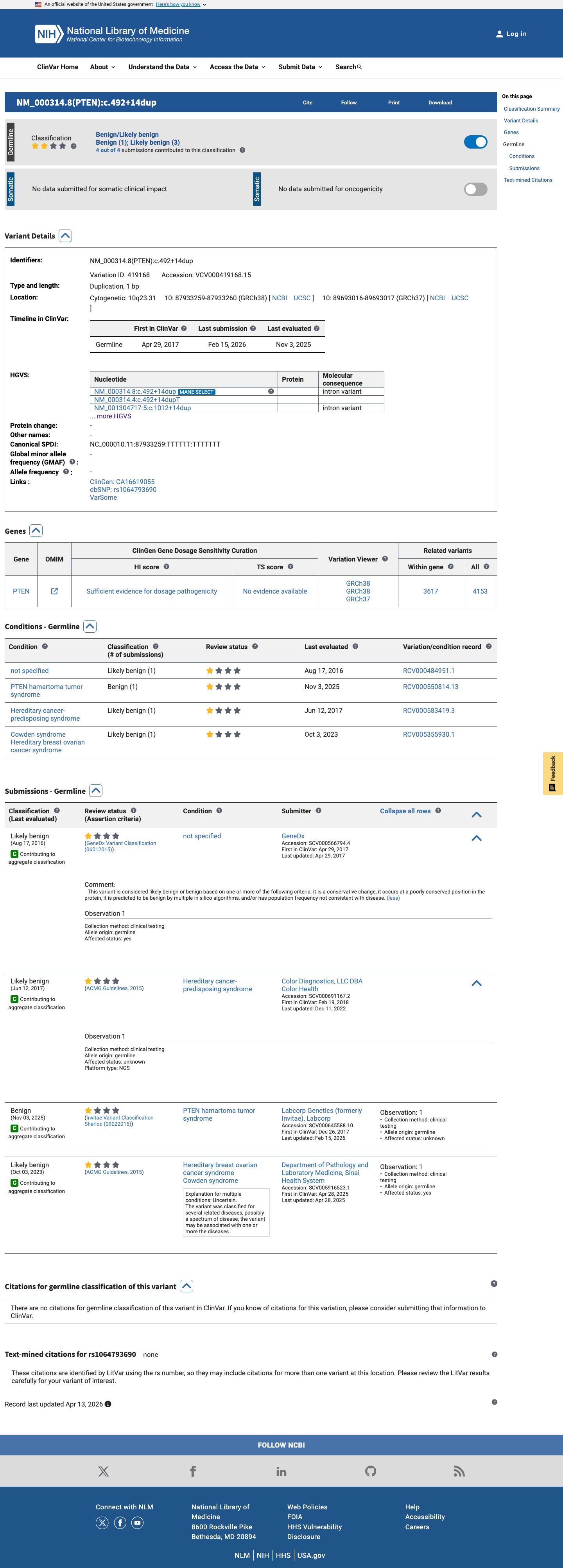
This variant has been reported in ClinVar as Likely benign (3 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 419168)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
23918944 ↗
Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
12692171 ↗
American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
17392385 ↗
American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography.
CLINVAR
17508274 ↗
Risk assessment and genetic counseling for hereditary breast and ovarian cancer: recommendations of the National Society of Genetic Counselors.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR