NM_000368.5:c.2194C>T (p.His732Tyr) is a missense variant in TSC1, a gene in which loss-of-function variants cause tuberous sclerosis complex (autosomal dominant). The variant is present in gnomAD v2.1 at an allele frequency of 0.387% (1093/282512 alleles) with 7 homozygotes, and in gnomAD v4.1 at 0.360% (5806/1612416 alleles) with 28 homozygotes. This frequency exceeds the 0.3% threshold for BS1 (strong benign).1 The presence of 28 homozygous individuals in gnomAD v4.1 is incompatible with a highly penetrant autosomal dominant tumor suppressor disorder; homozygous loss of TSC1 function would be expected to cause severe disease.2 Multiple in silico predictors support a benign interpretation: REVEL score 0.338 (below 0.5 pathogenic threshold), BayesDel score -0.064 (negative score), and SpliceAI max delta 0.09 (below 0.2 splicing threshold), meeting BP4 (supporting benign).3 In ClinVar, 23 clinical laboratories classify this variant as Benign and 4 as Likely benign (Variation ID: 5103), meeting BP6 (supporting benign).4 PMID:19918125 (Lugnier et al. 2009) characterized hamartin H732Y in vitro and demonstrated reduced tuberin binding (2.3-fold, p<0.05) and aberrant nuclear localization. However, the paper explicitly reported the allele at 0.5% frequency in the normal population and concluded it 'cannot be sufficient to cause TSC or FCD,' describing it as a low-penetrance predisposing variant at most.5 Applying generic ACMG/AMP 2015 combination rules (PMID:25741868): one strong benign criterion (BS1) plus two supporting benign criteria (BP4, BP6) yields a final classification of Likely Benign.6
TSC1
Final classification
Likely Benign
TSC1 c.2194C>T · p.His732Tyr
TSC1
NM_000368.5:c.2194C>T (p.His732Tyr) is a missense variant in TSC1, a gene in which loss-of-function variants cause tuberous sclerosis complex (autosomal dominant).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong, BP4 supporting benign, BP6 supporting benign; combination = 1 strong benign + 2 supporting benign, which maps to Likely Benign.
Classification rationale
BS1BP4BP6
Likely Benign
TSC1 c.2194C>T
BS1 + BP4 + BP6
→
Likely Benign
3
revelbayesdelspliceai ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_000368.5 · variants mapped to exon structure
TSC1
NM_000368.5
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 17 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
The variant is present in gnomAD v2.1 at AF 0.387% (1093/282512 alleles, 7 homozygotes) and gnomAD v4.1 at AF 0.360% (5806/1612416 alleles, 28 homozygotes). Both overall frequencies exceed the 0.3% BS1 threshold. The presence of 7–28 apparently healthy homozygous individuals in population databases for an autosomal dominant tumor suppressor gene (TSC1) provides strong evidence that this variant is benign. PMID:19918125 independently reported the allele at 0.5% in the normal population and concluded it 'cannot be sufficient to cause TSC or FCD.'
gnomAD v2.1 AF=0.387% with 7 homozygotesgnomAD v4.1 AF=0.360% with 28 homozygotesPMID:19918125 reports 0.5% in normal population and explicitly states variant cannot cause TSC
✓
BP4
supporting
Benign
Multiple in silico predictors consistently indicate a benign impact. REVEL score is 0.338 (below the 0.5 threshold for pathogenic prediction), BayesDel score is -0.064 (negative, favoring benign), and SpliceAI max delta is 0.09 (below the 0.2 threshold for splicing impact). All three orthogonal computational tools agree on a benign/non-damaging prediction.
REVEL=0.338 (<0.5)BayesDel=-0.064 (negative score)SpliceAI delta=0.09 (<0.2)
✓
BP6
supporting
Benign
This variant is classified as Benign in ClinVar by 23 clinical laboratories and as Likely benign by 4 clinical laboratories (ClinVar Variation ID: 5103). The review status is 'criteria provided, single submitter.' The strong clinical consensus — 23 out of 27 classified submissions are 'Benign' — supports a benign interpretation under BP6.
ClinVar: Benign (23 labs)Likely benign (4 labs)Variation ID 5103
Assessed · not applied
Pathogenic
PS1
No known pathogenic variant resulting in the same amino acid change (His732Tyr) was identified.
PS2
One confirmed de novo occurrence reported (PMID:17304050) in a TSC proband with confirmed parental testing, but PS2 requires ≥2 de novo observations for strong evidence.
PS3
PMID:19918125 (Lugnier et al.
PS4
The variant has been observed in TSC patient cohorts (1/325 probands in PMID:17304050; also reported in PMID:15798777), but the prevalence in affected individuals does not significantly exceed the general population frequency of ~0.37% (gnomAD).
PM1
Residue His732 lies outside the well-established TSC1 functional domains.
PM2
The variant is present in gnomAD v2.1 at AF 0.00387 (0.387%, 1093/282512 alleles, 7 homozygotes) and in gnomAD v4.1 at AF 0.00360 (0.360%, 5806/1612416 alleles, 28 homozygotes).
PM6
One presumed de novo occurrence without confirmation of paternity/maternity was reported (PMID:15798777).
PP1
No published cosegregation data are available for this variant in families with multiple affected individuals.
PP3
Multiple in silico predictors suggest a benign impact: REVEL score 0.338 (threshold ≥0.5 for pathogenic), BayesDel score -0.064 (threshold >0 for pathogenic), SpliceAI max delta 0.09 (threshold ≥0.2 for splice effect).
PP4
PP4 requires detailed patient phenotype information and specificity scoring; insufficient phenotype data are available to assess whether the variant is enriched in patients with a phenotype highly specific for TSC1-related disease.
PP5
ClinVar classification for this variant is Benign (23 clinical laboratories) and Likely benign (4 laboratories); PP5 is applicable only when reputable sources classify the variant as pathogenic.
Benign
BA1
The overall gnomAD allele frequency is 0.37% (v2.1) and 0.36% (v4.1), which is below the 1% BA1 threshold.
BS2
BS2 (observed in a healthy adult individual for a fully penetrant disorder) is superseded by BS1, which provides stronger population-based evidence using allele frequency rather than individual observations.
BS3
No well-established functional studies demonstrate that the H732Y variant has no damaging effect on protein function.
BS4
No published reports demonstrate lack of segregation of this variant with disease in affected families.
BP1
BP1 is applicable to missense variants in genes where primarily truncating variants cause disease.
BP5
BP5 requires an alternate molecular basis for disease in a case where the variant is observed.
N/A · 6
PVS1 · PM5 · PP2 · BP2 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
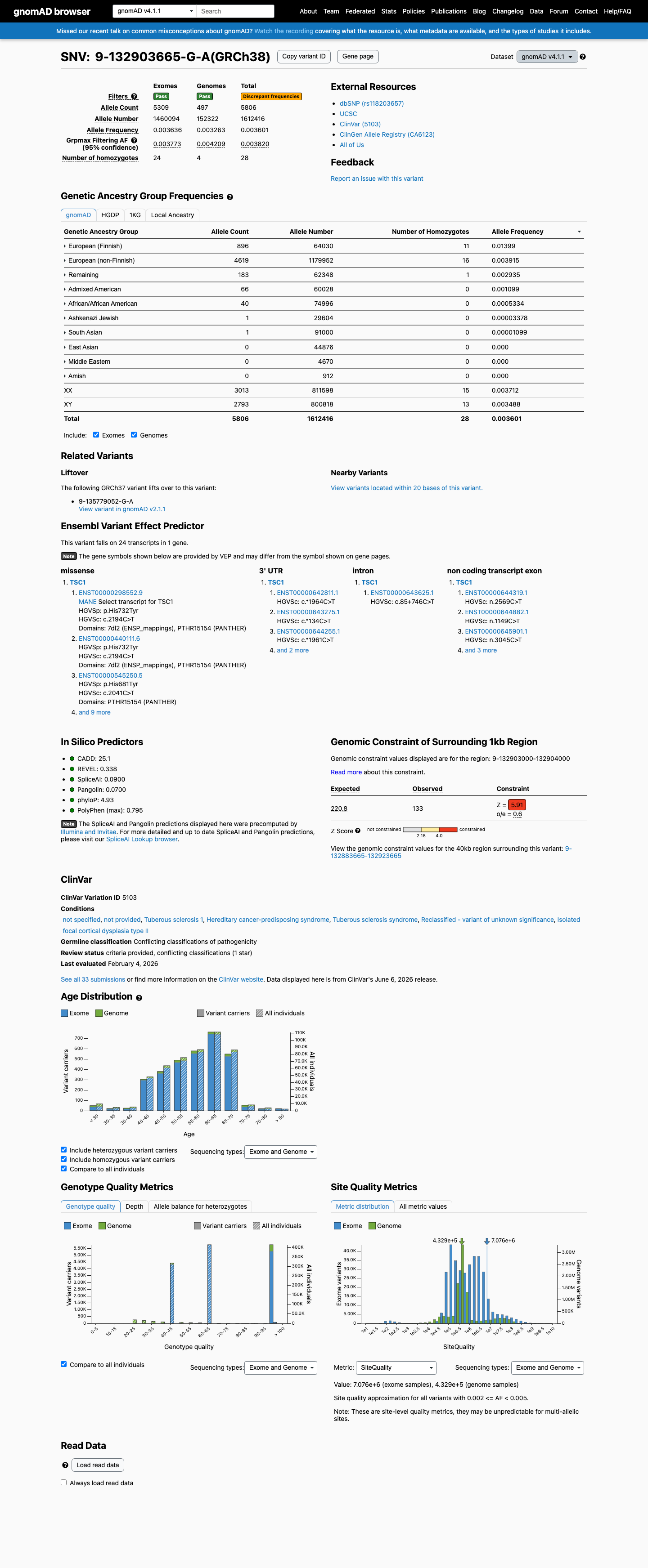
This variant is present in gnomAD v4.1 (AF= 0.00360081; MAF= 0.36008%, 5806/1612416 alleles, homozygotes = 28) and has highest observed frequency in the European (Finnish) population (AF= 0.0139934; MAF= 1.39934%, 896/64030 alleles, homozygotes = 11); grpmax FAF= 0.00382016.
v2.1
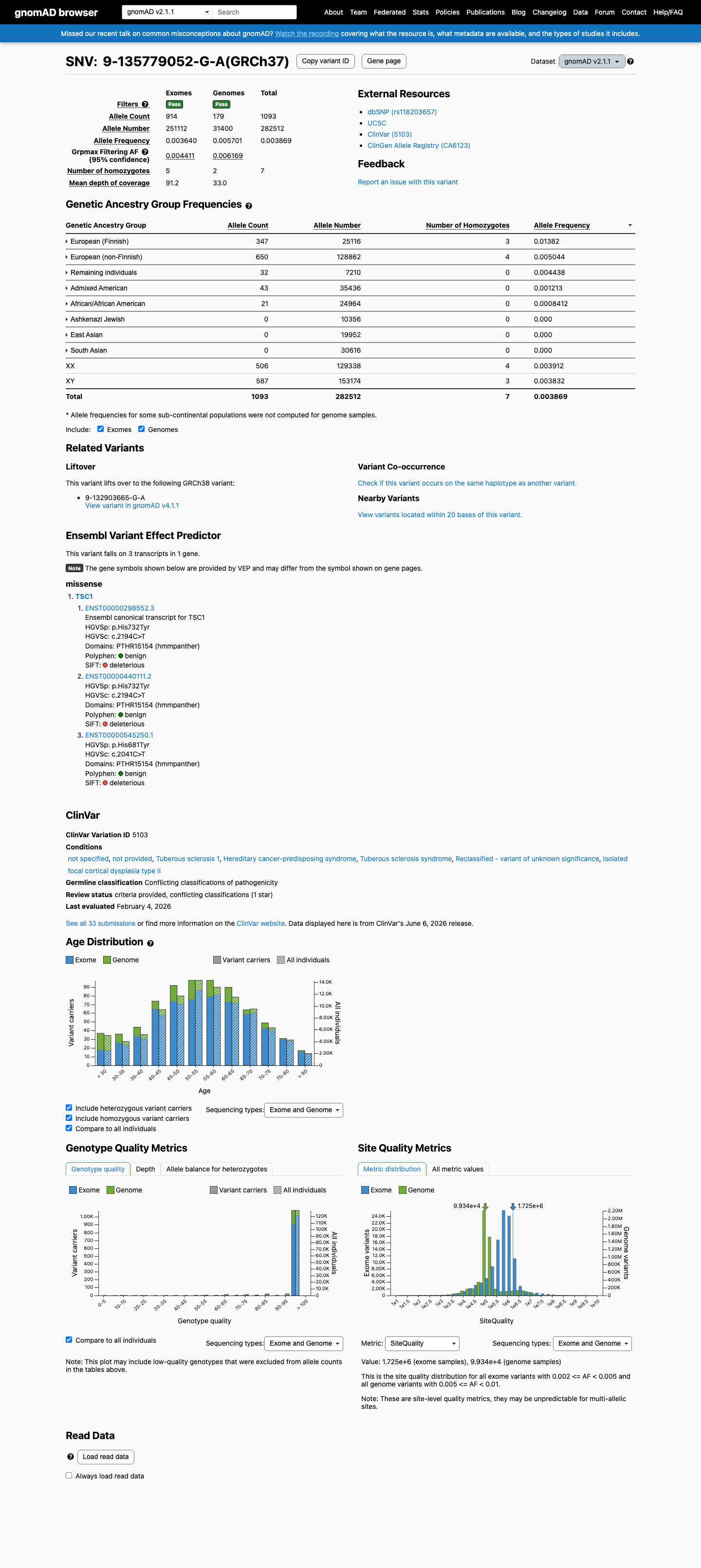
This variant is present in gnomAD v2.1 (AF= 0.00386886; MAF= 0.38689%, 1093/282512 alleles, homozygotes = 7) and has highest observed frequency in the European (Finnish) population (AF= 0.0138159; MAF= 1.38159%, 347/25116 alleles, homozygotes = 3); grpmax FAF= 0.00616893.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.002062975027144408, 38/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.36%
· 5806 / 1,612,416
28 hom · FAF 0.38%
28 hom · FAF 0.38%
European (Finnish) 896 / 64,030 |
1.4% 11 hom |
European (non-Finnish) 4619 / 1,179,952 |
0.39% 16 hom |
Remaining individuals 183 / 62,348 |
0.29% 1 hom |
Admixed American 66 / 60,028 |
0.11% |
African/African American 40 / 74,996 |
0.053% |
Ashkenazi Jewish 1 / 29,604 |
0.0034% |
South Asian 1 / 91,000 |
0.0011% |
+ 3 not observed (Amish, East Asian, Middle Eastern)
gnomAD v2.1
0.39%
· 1093 / 282,512
7 hom · FAF 0.62%
7 hom · FAF 0.62%
European (Finnish) 347 / 25,116 |
1.4% 3 hom |
European (non-Finnish) 650 / 128,862 |
0.5% 4 hom |
Remaining individuals 32 / 7,210 |
0.44% |
Admixed American 43 / 35,436 |
0.12% |
African/African American 21 / 24,964 |
0.084% |
+ 3 not observed (Ashkenazi Jewish, East Asian, South Asian)
gnomAD Canada 🇨🇦
0.21%
· 38 / 18,420
0 hom · FAF 0.22%
0 hom · FAF 0.22%
European (non-Finnish) 35 / 11,742 |
0.3% |
African/African American 2 / 1,020 |
0.2% |
Remaining individuals 1 / 1,138 |
0.088% |
+ 6 not observed (Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, South Asian)

ClinVar
This variant has been reported in ClinVar as Benign (23 clinical laboratories) and as Likely benign (4 clinical laboratories). (ClinVarID = 5103)
In silico
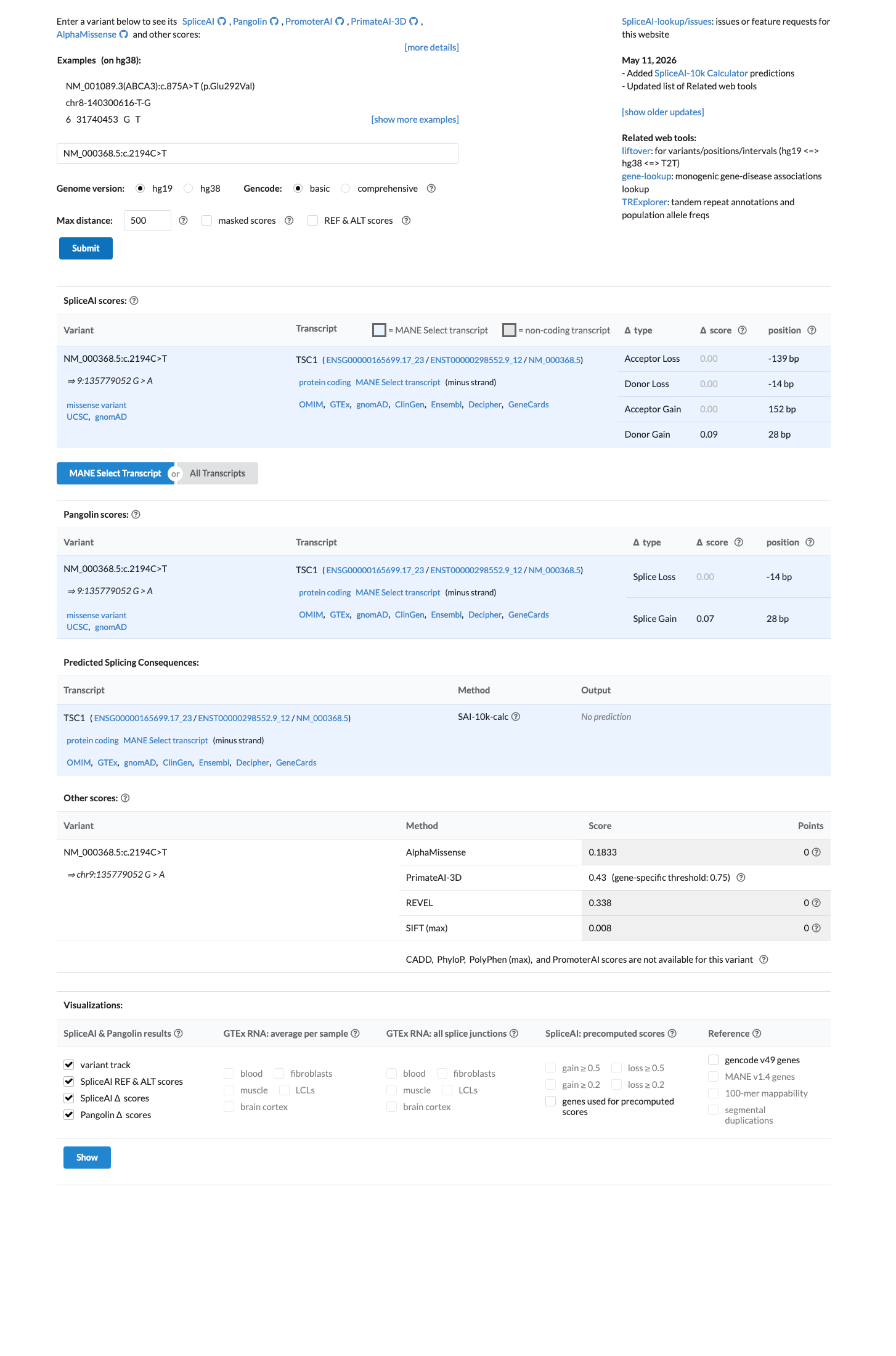
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.09). REVEL score = 0.338. BayesDel score = -0.0641334.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. TSC1, a scaffold protein, is frequently altered by mutation in bladder cancer.
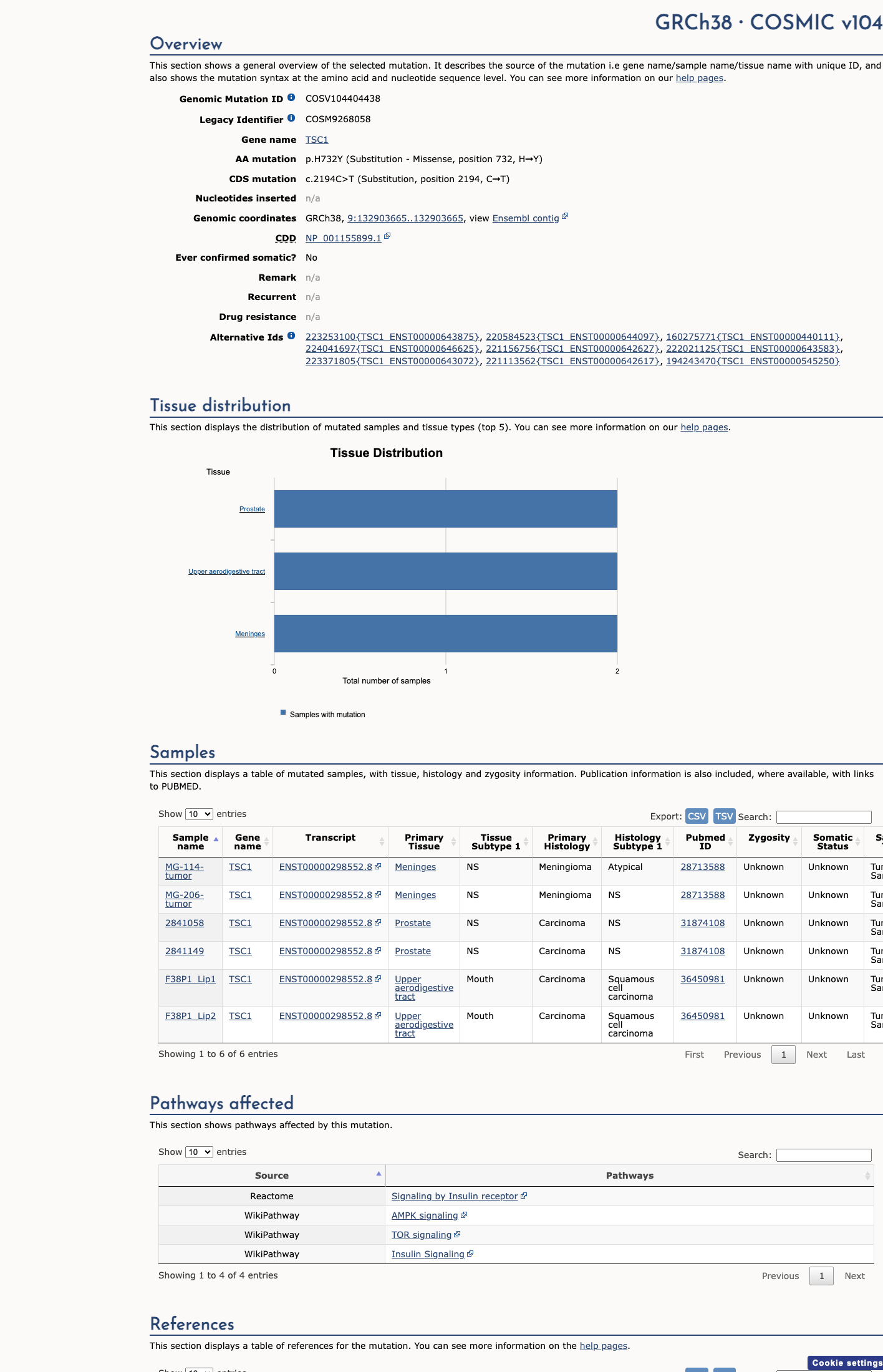
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV104404438, n = 6 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 9 further PMIDs triaged but not cited — see Sources & References.
Hamartin variants that are frequent in focal dysplasias and cortical tubers have reduced tuberin binding and aberrant subcellular distribution in vitro.
Found
gnomAD v2.1 AF=0.387% with 7 homozygotes gnomAD v4.1 AF=0.360% with 28 homozygotes PMID:19918125 reports 0.5% in normal population and explicitly states variant cannot cause TSC
Applied to
→BS1 supports · met
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
10227394 ↗
Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation.
CLINVAR
21309039 ↗
Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with Tuberous Sclerosis Complex.
CLINVAR
23514105 ↗
Lack of association of rare functional variants in TSC1/TSC2 genes with autism spectrum disorder.
CLINVAR
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
27425891 ↗
Minute amounts of hamartin wildtype rescue the emergence of tuber-like lesions in conditional Tsc1 ablated mice.
CLINVAR
18414213 ↗
ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR