NM_000455.5:c.*9G>A is a 3' UTR variant in STK11 located 9 nucleotides downstream of the stop codon. The variant is absent from gnomAD v2.1 (0/109,868 alleles) and present at extremely low frequency in gnomAD v4.1 (9/1,511,712 alleles; AF=0.00006%), supporting application of PM2 at supporting strength.1 ClinVar lists this variant under ID 387684 with mixed classifications (Likely benign from 3 laboratories, Uncertain significance from 3 laboratories, Benign from 1 laboratory) at 1-star review status, which does not meet the 3-star threshold for PP5 or BP6.2 No functional studies, segregation data, de novo observations, case-control data, or phenotype information are available for this variant. No published literature mentions NM_000455.5:c.*9G>A specifically. SpliceAI predicts no splicing impact (max delta score = 0.00), consistent with a benign splicing effect, though insufficient alone to meet BP4.3 With only one pathogenic criterion met at supporting strength (PM2) and no benign criteria met, the evidence is insufficient to classify this variant beyond Variant of Uncertain Significance (VUS) per ACMG/AMP 2015 combination rules.4
STK11
Final classification
VUS
STK11 c.*9G>A · p.?
STK11
NM_000455.5:c.*9G>A is a 3' UTR variant in STK11 located 9 nucleotides downstream of the stop codon.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting; combination = 1 supporting, which maps to VUS.
Classification rationale
PM2
VUS
STK11 c.*9G>A
PM2
→
VUS
Gene diagram
· NM_000455.5 · variants mapped to exon structure
STK11
NM_000455.5
Fetching transcript structure from UCSC…
Applied criteria · 1 applied · 17 assessed
Applied · 1
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000455.5:c.*9G>A is absent from gnomAD v2.1 (0/109,868 alleles) and present at an extremely low frequency in gnomAD v4.1 (9/1,511,712 alleles; AF=0.00006%), well below the 0.1% threshold for PM2. The variant is also absent from gnomAD-Canada v1.0.
gnomAD v2.1: 0/109868 alleles (AF=0%)gnomAD v4.1: 9/1
Assessed · not applied
Pathogenic
PS2
No de novo observation has been reported for NM_000455.5:c.*9G>A in any reviewed source.
PS3
No functional studies have been performed on NM_000455.5:c.*9G>A.
PS4
No case-control or prevalence data are available comparing the frequency of NM_000455.5:c.*9G>A in affected individuals versus controls.
PM1
The variant is located in the 3' UTR (c.*9), 9 nucleotides downstream of the stop codon.
PM6
No de novo observation of NM_000455.5:c.*9G>A with confirmed parentage has been reported.
PP1
No cosegregation data are available for NM_000455.5:c.*9G>A.
PP3
No in silico predictors support a damaging effect.
PP4
No patient phenotype or clinical data are available for individuals carrying NM_000455.5:c.*9G>A.
PP5
ClinVar reports NM_000455.5:c.*9G>A as Likely benign (3 labs), Uncertain significance (3 labs), and Benign (1 lab) under variation ID 387684.
Benign
BA1
The maximum allele frequency of NM_000455.5:c.*9G>A in gnomAD is 0.00006% (gnomAD v4.1, 9/1,511,712 alleles), far below the 1% threshold required for BA1.
BS1
The allele frequency of NM_000455.5:c.*9G>A is 0.00006% (gnomAD v4.1), far below the 0.3% threshold required for BS1.
BS2
NM_000455.5:c.*9G>A has not been observed in a homozygous state in gnomAD (0 homozygotes in both v2.1 and v4.1).
BS3
No functional studies demonstrating a benign effect have been performed on NM_000455.5:c.*9G>A.
BS4
No segregation data are available for NM_000455.5:c.*9G>A to evaluate lack of segregation with disease.
BP2
No data are available regarding observation of NM_000455.5:c.*9G>A in trans with a known pathogenic STK11 variant.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact.
BP6
ClinVar reports NM_000455.5:c.*9G>A as Likely benign (3 labs), Uncertain significance (3 labs), and Benign (1 lab).
N/A · 7
PVS1 · PS1 · PM5 · PP2 · BP1 · BP5 · BP7
Research & evidence
Population frequency
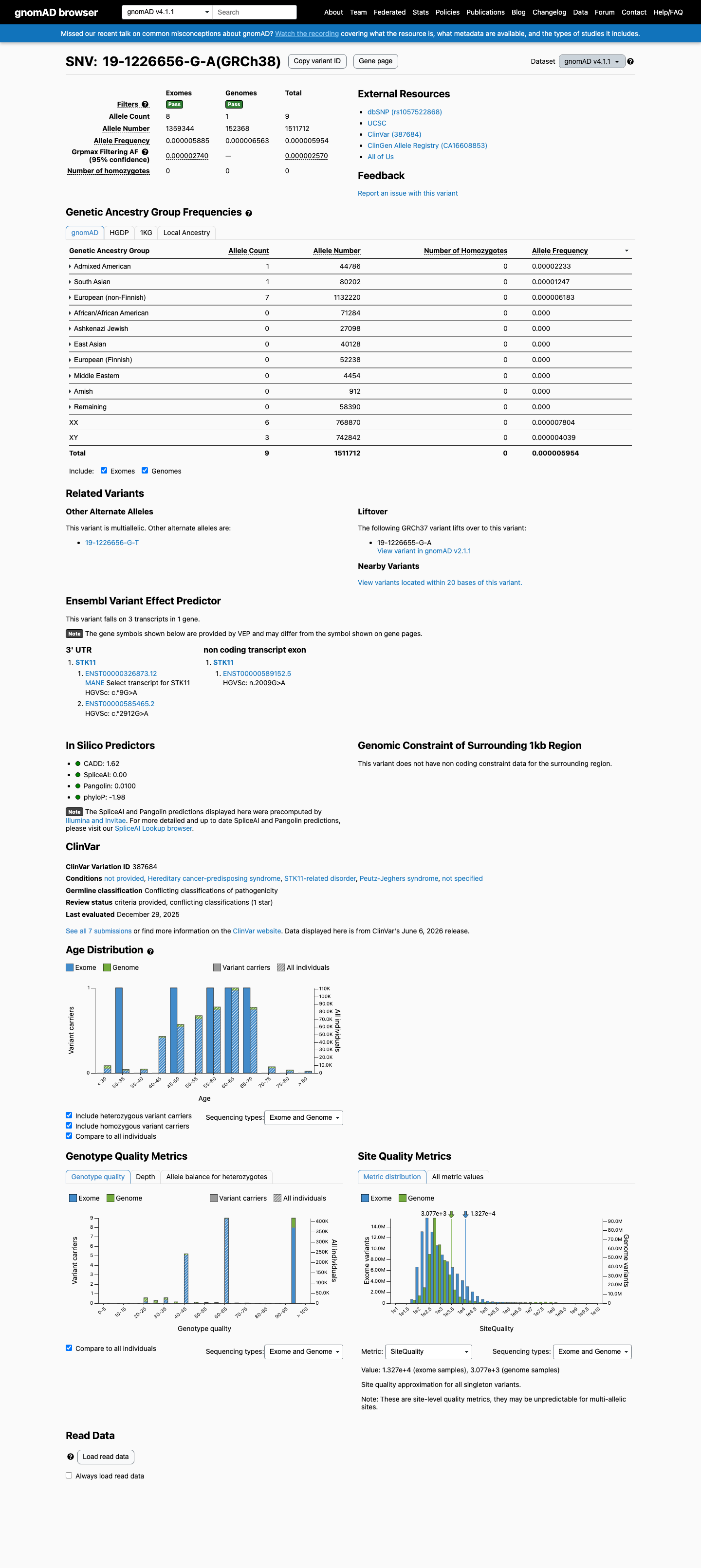
gnomAD v4.1
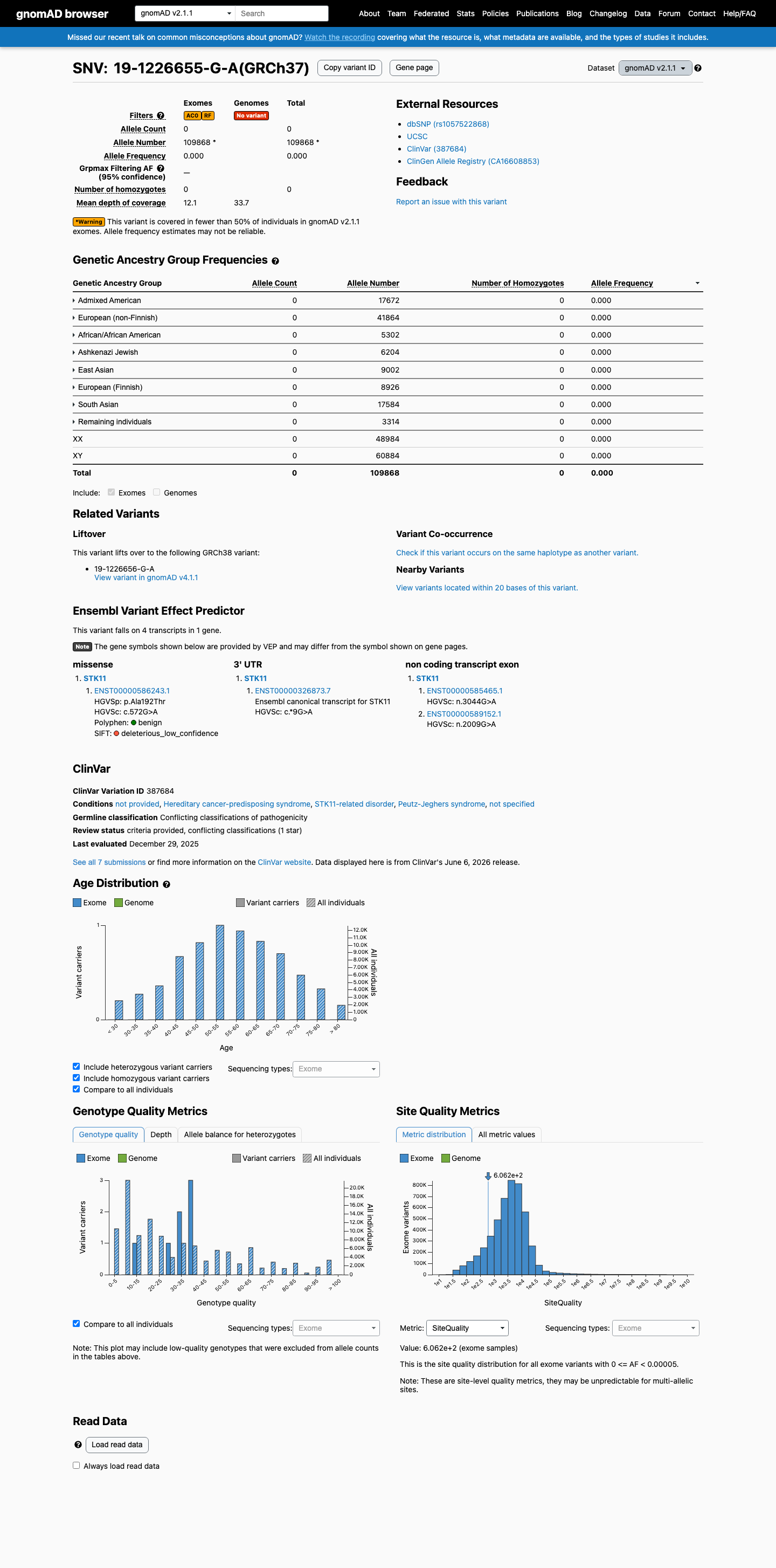
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 5.95351e-06; MAF= 0.00060%, 9/1511712 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 2.23284e-05; MAF= 0.00223%, 1/44786 alleles, homozygotes = 0); grpmax FAF= 2.57e-06.
v2.1
This variant is present in gnomAD v2.1 (AF= 0; MAF= 0.00000%, 0/109868 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/5302 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0006%
· 9 / 1,511,712
0 hom · FAF 0.00026%
0 hom · FAF 0.00026%
Admixed American 1 / 44,786 |
0.0022% |
South Asian 1 / 80,202 |
0.0012% |
European (non-Finnish) 7 / 1,132,220 |
0.00062% |
+ 7 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / 109,868
0 hom
0 hom
Not observed in any ancestry group.
+ 8 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
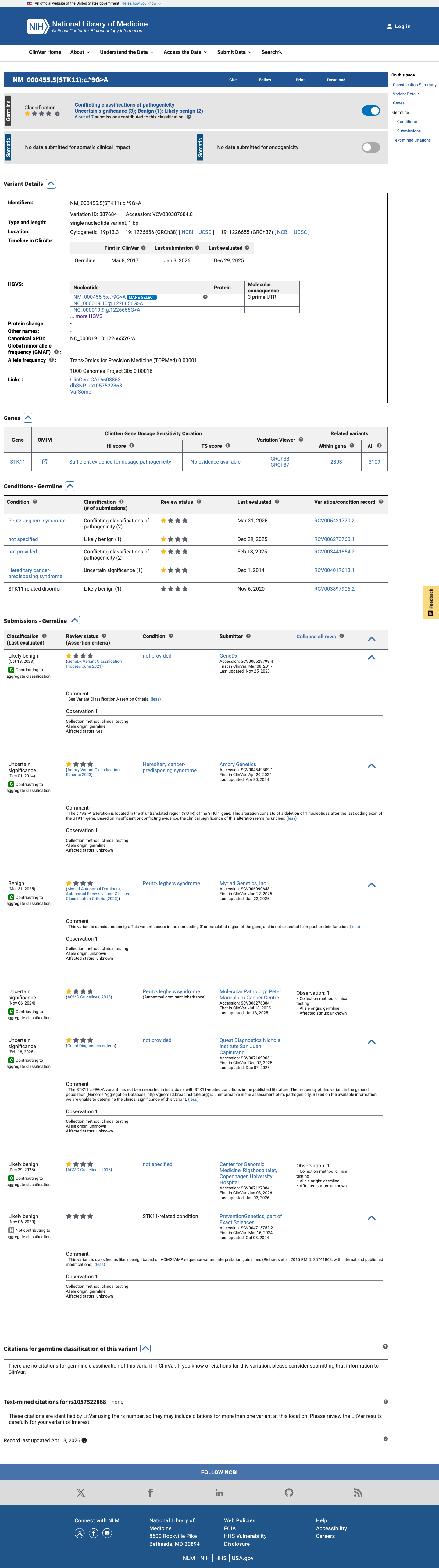
ClinVar
This variant has been reported in ClinVar as Likely benign (3 clinical laboratories) and as Uncertain significance (3 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 387684)
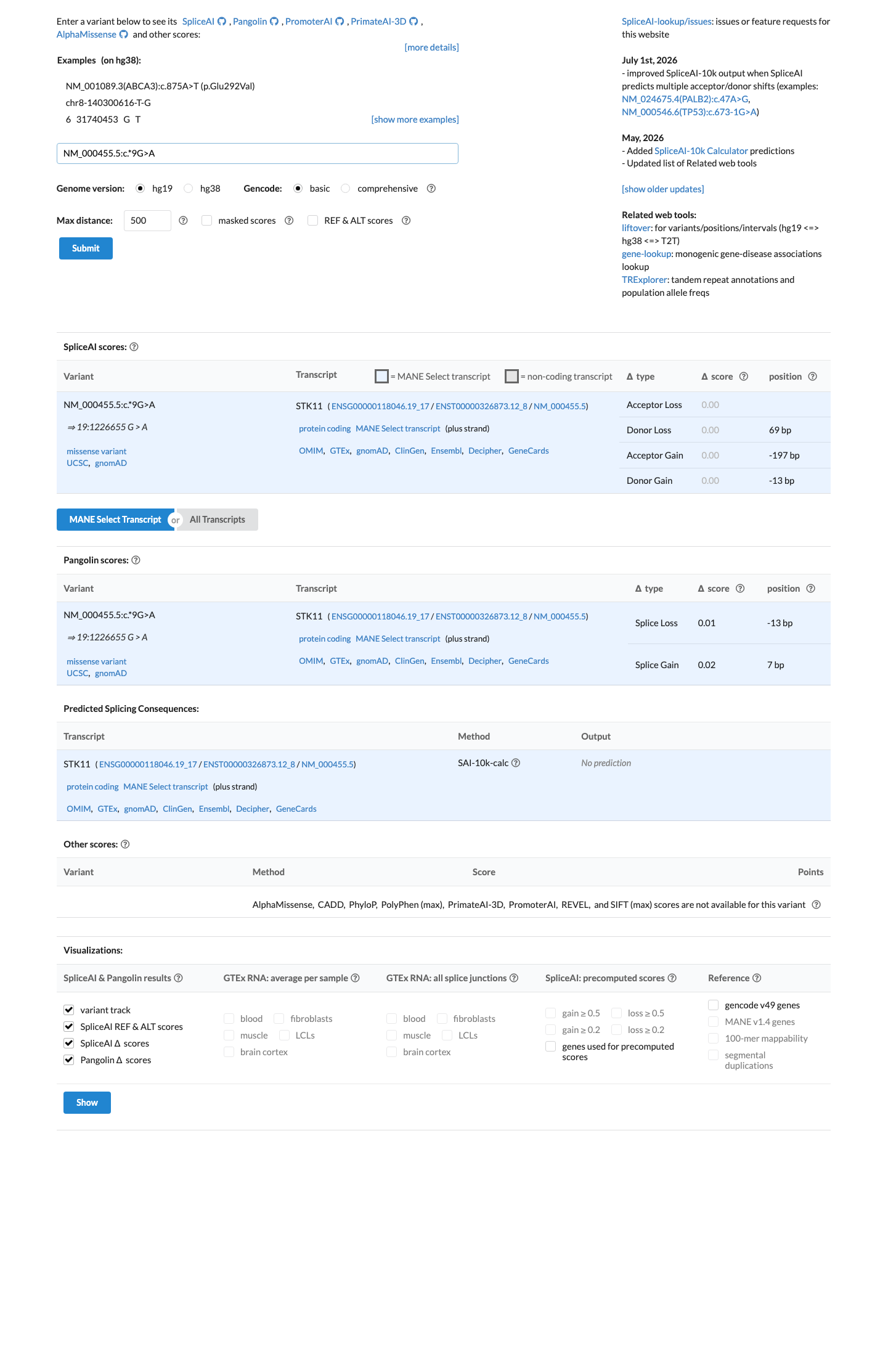
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
Somatic evidence
COSMIC
This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant cancer hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25645574 ↗
ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
26389258 ↗
Cancer Genetics Risk Assessment and Counseling (PDQ®): Health Professional Version.
CLINVAR
27854360 ↗
Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics.
CLINVAR