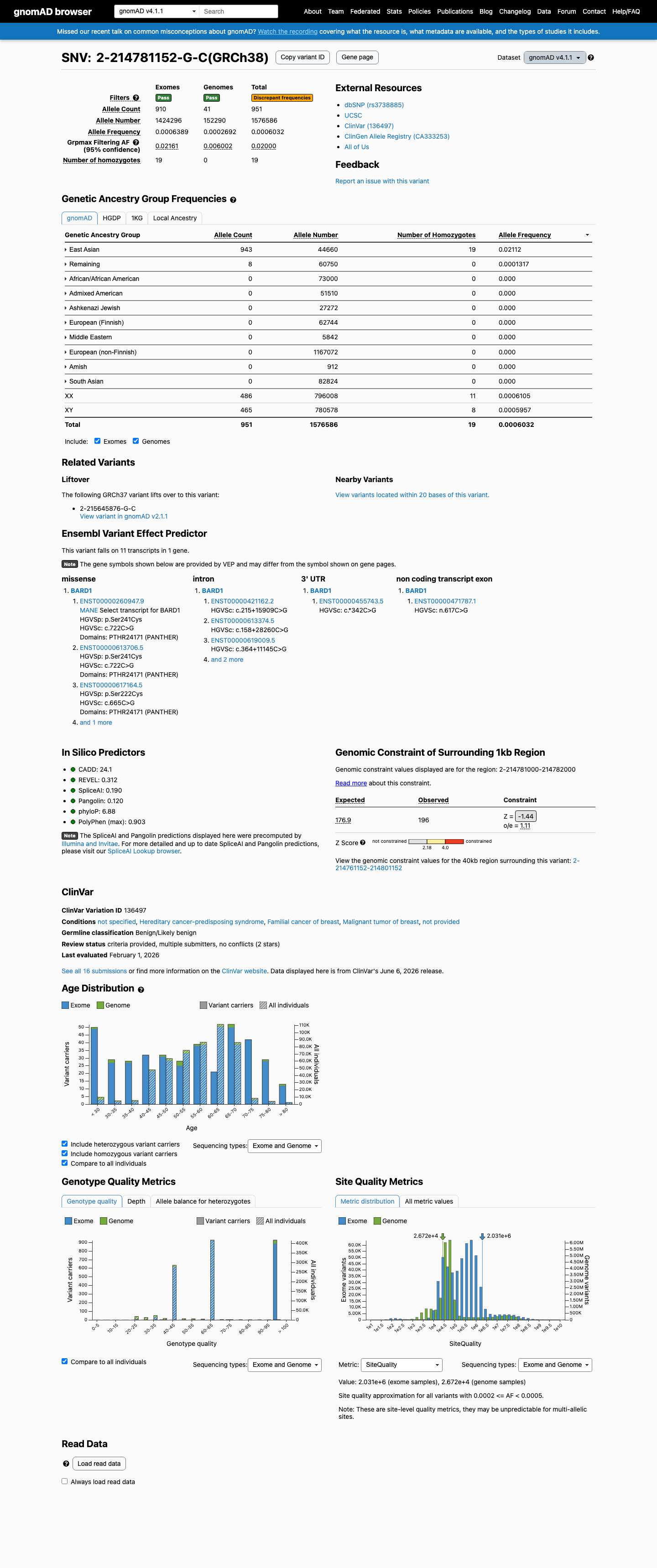
This variant is present in gnomAD v4.1 at an East Asian allele frequency of 2.11% (943/44,660 alleles) with 19 homozygotes and a grpmax filtering allele frequency of 2.0%, exceeding the 1% BA1 stand-alone benign threshold and establishing it as a common polymorphism incompatible with a highly penetrant Mendelian disorder.1 The variant is classified as Benign in ClinVar (Variation ID 136497) with consensus from 9 clinical testing laboratories reporting Benign and 6 reporting Likely benign.2 Multiple in silico predictors are consistent with a benign effect: REVEL score 0.312, BayesDel score -0.180, and SpliceAI maximum delta score 0.19 indicate no significant impact on protein function or splicing.3 The variant was listed among 11 BARD1 nsSNPs predicted as 'not tolerated' by SIFT (TI=0.04) in an in silico screening study (PMID:23056176), but was not classified as damaging by PolyPhen and was not among the four SNPs identified as deleterious by both methods. This in silico analysis does not alter the benign classification.4 No variant-specific functional studies, de novo reports, or segregation data were identified. The variant has not been reported as pathogenic by any reputable source.5 Final classification: Benign. BA1 (stand-alone benign) is met. BS1 (strong benign) and BP4, BP6 (supporting benign) provide additional support. No pathogenic criteria are met.6
BARD1
Final classification
Benign
BARD1 c.722C>G · p.Ser241Cys
BARD1
This variant is present in gnomAD v4.1 at an East Asian allele frequency of 2.11% (943/44,660 alleles) with 19 homozygotes and a grpmax filtering allele frequency of 2.0%, exceeding the 1% BA1 stand-alone benign threshold and establishing it as a common polymorphism incompatible with a highly penetrant Mendelian disorder.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BA1 stand-alone benign, BS1 strong, BP4 supporting, BP6 supporting; combination = 1 stand-alone benign + 1 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BA1BS1BP4BP6
Benign
BARD1 c.722C>G
BA1 + BS1 + BP4 + BP6
→
Benign
Gene diagram
· NM_000465.4 · variants mapped to exon structure
BARD1
NM_000465.4
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 18 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
gnomAD v4.1 reports an East Asian allele frequency of 2.11% (943/44,660 alleles) with 19 homozygotes, and a grpmax filtering allele frequency of 2.0%. These exceed the 1% BA1 threshold, establishing this variant as a common polymorphism incompatible with a highly penetrant Mendelian disease.
gnomAD v4.1: EAS AF 2.11% (943/4466019 homozygotes)
✓
BS1
strong
Benign
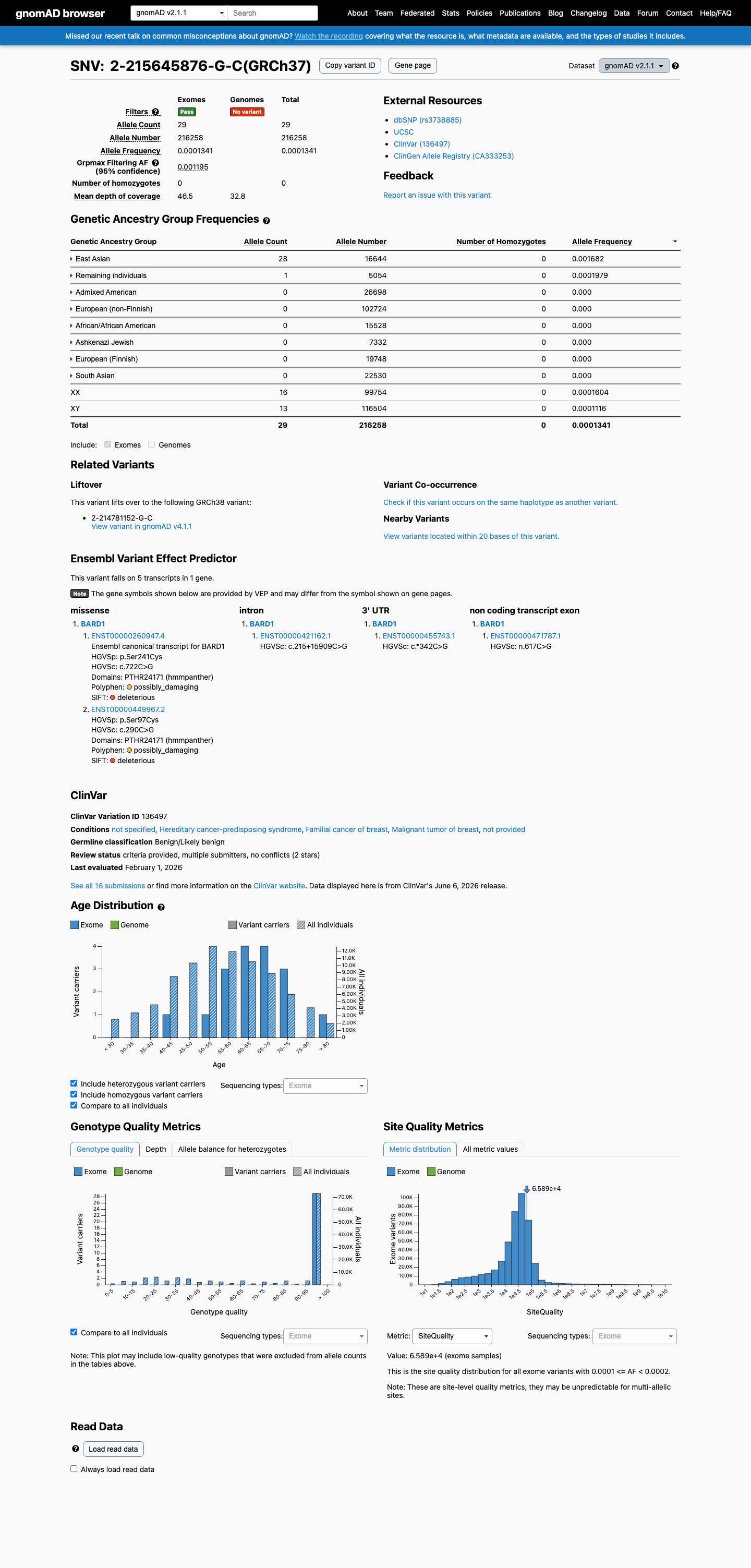
gnomAD v4.1 grpmax filtering allele frequency of 2.0% and East Asian allele frequency of 2.11% far exceed the 0.3% BS1 threshold. gnomAD v2.1 grpmax FAF of 0.12% is below 0.3% but superseded by the larger v4.1 dataset.
gnomAD v4.1 grpmax FAF 2.0% and EAS AF 2.11% exceed 0.3% BS1 threshold.
✓
BP4
supporting
Benign
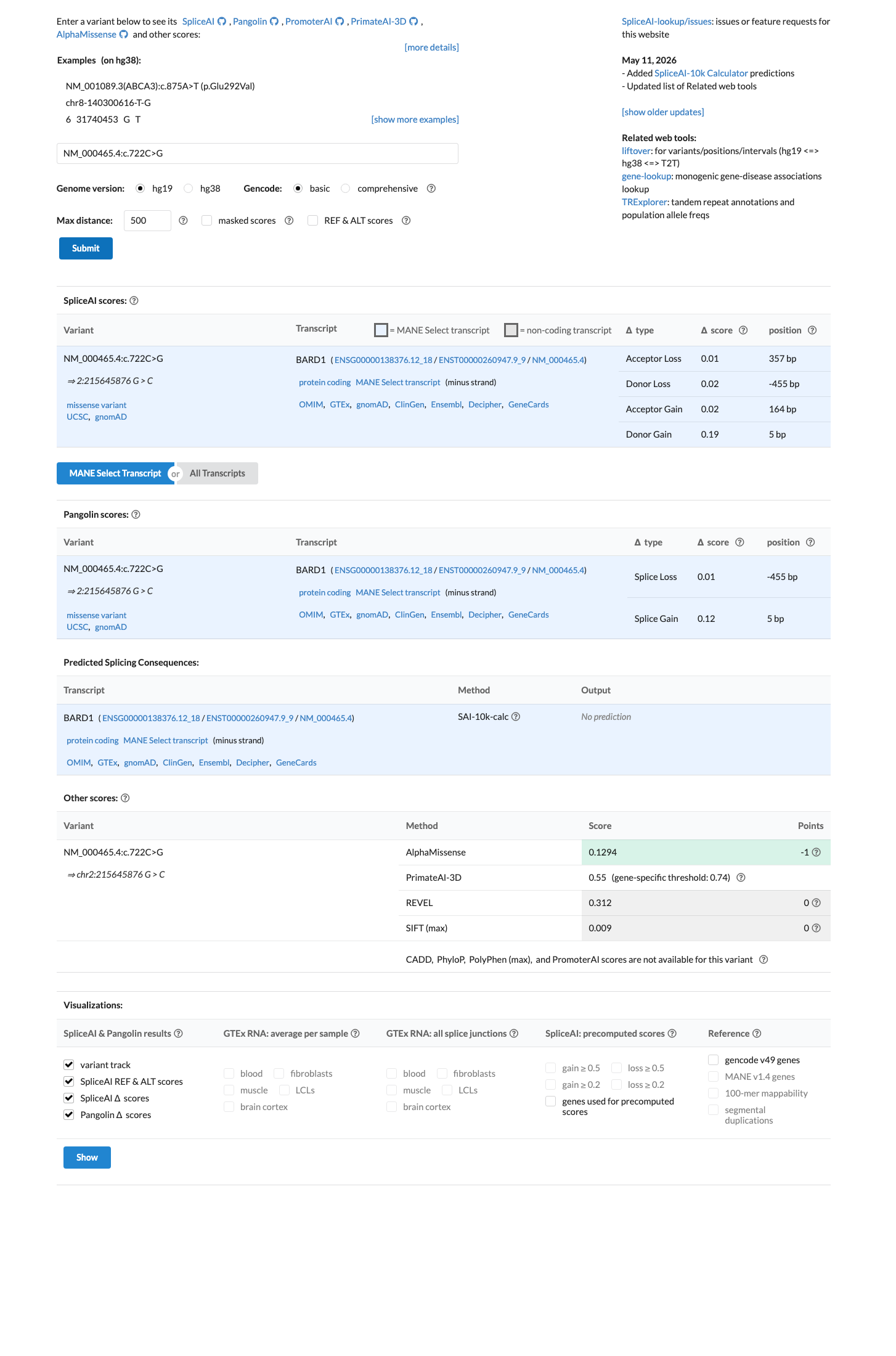
Multiple lines of computational evidence suggest no significant impact on the gene product. REVEL score is 0.312 (below pathogenic threshold), BayesDel score is -0.180 (benign range), and SpliceAI max delta score is 0.19 (no predicted splicing impact). Convergent benign predictions from independent in silico tools meet BP4 at supporting level.
REVEL 0.312BayesDel -0.180SpliceAI max delta 0.19. Three independent computational methods consistently predict no significant deleterious effect.
✓
BP6
supporting
Benign
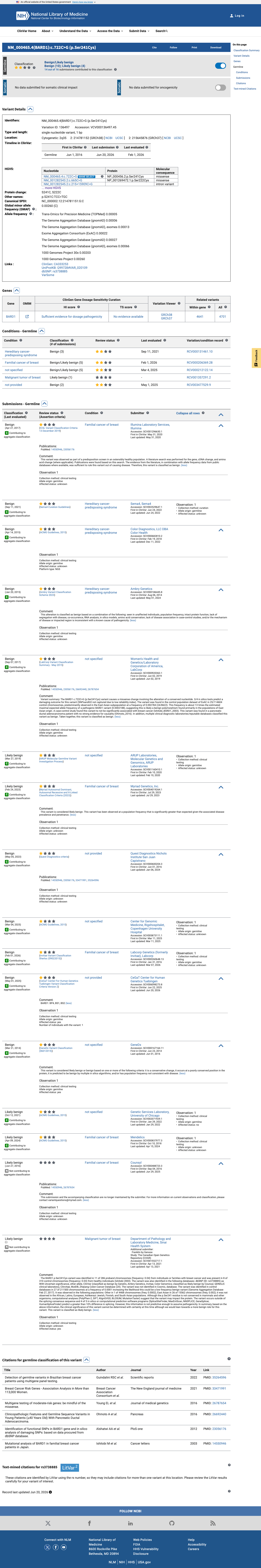
ClinVar reports this variant as Benign by consensus of 9 clinical laboratories and Likely benign by 6 additional laboratories (ClinVar Variation ID 136497). The aggregate classification from multiple clinical testing laboratories is Benign, meeting BP6 at supporting level.
ClinVar: Benign (9 labs)Likely benign (6 labs). Aggregate classification Benign. ClinVar ID 136497.
Assessed · not applied
Pathogenic
PS1
No previously established pathogenic variant resulting in the same amino acid change (p.Ser241Cys) via a different nucleotide change was identified.
PS2
No de novo occurrence of this variant was reported in any reviewed source.
PS3
No well-established in vitro or in vivo functional studies were identified for this variant.
PS4
The variant is observed at high frequency in population databases (gnomAD v4.1 East Asian AF 2.11%, 19 homozygotes), indicating it is a common polymorphism.
PM1
The variant does not lie in a statistically significant mutational hotspot.
PM2
gnomAD grpmax filtering allele frequency of 0.12% (v2.1) and 2.0% (v4.1) exceeds the 0.1% threshold for PM2.
PM6
No de novo report was identified for this variant.
PP1
No segregation data were identified for this variant.
PP2
PP2 applies to genes with a low rate of benign missense variation where missense variants are a common disease mechanism.
PP3
Multiple lines of computational evidence do not support a deleterious effect.
PP4
No specific patient phenotype or family history consistent with BARD1-related hereditary breast and ovarian cancer syndrome was provided for assessment.
PP5
No reputable source reports this variant as pathogenic.
Benign
BS2
BS2 requires observation in a healthy adult individual for a disorder with full penetrance expected at an early age.
BS3
No well-established in vitro or in vivo functional studies demonstrating a neutral effect were identified.
BS4
No segregation data were available to assess lack of cosegregation with disease in affected family members.
BP1
BP1 applies to missense variants in genes for which primarily truncating variants are known to cause disease.
BP2
No evidence was found of this variant occurring in trans with a known pathogenic BARD1 variant in a fully penetrant dominant disorder.
BP5
No alternate molecular basis for disease was identified in a case harboring this variant.
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000603202; MAF= 0.06032%, 951/1576586 alleles, homozygotes = 19) and has highest observed frequency in the East Asian population (AF= 0.0211151; MAF= 2.11151%, 943/44660 alleles, homozygotes = 19); grpmax FAF= 0.0199964.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000134099; MAF= 0.01341%, 29/216258 alleles, homozygotes = 0) and has highest observed frequency in the East Asian population (AF= 0.00168229; MAF= 0.16823%, 28/16644 alleles, homozygotes = 0); grpmax FAF= 0.00119534.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00016286644951140066, 3/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.06%
· 951 / 1,576,586
19 hom · FAF 2%
19 hom · FAF 2%
East Asian 943 / 44,660 |
2.1% 19 hom |
Remaining individuals 8 / 60,750 |
0.013% |
+ 8 not observed (Admixed American, European (Finnish), Amish, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
0.013%
· 29 / 216,258
0 hom · FAF 0.12%
0 hom · FAF 0.12%
East Asian 28 / 16,644 |
0.17% |
Remaining individuals 1 / 5,054 |
0.02% |
+ 6 not observed (African/African American, Admixed American, Ashkenazi Jewish, European (Finnish), European (non-Finnish), South Asian)
gnomAD Canada 🇨🇦
0.016%
· 3 / 18,420
0 hom · FAF 0.027%
0 hom · FAF 0.027%
East Asian 2 / 1,338 |
0.15% |
Remaining individuals 1 / 1,138 |
0.088% |
+ 7 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, European (Finnish), Middle Eastern, European (non-Finnish), South Asian)
ClinVar
This variant has been reported in ClinVar as Benign (9 clinical laboratories) and as Likely benign (6 clinical laboratories). (ClinVarID = 136497)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.19). REVEL score = 0.312. BayesDel score = -0.180224.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. BARD1, a tumor suppressor involved in the DNA damage response, is altered by mutation in breast and ovarian cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
23056176 ↗
Identification of functional SNPs in BARD1 gene and in silico analysis of damaging SNPs: based on data procured from dbSNP database.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26467025 ↗
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders.
CLINVAR
26692440 ↗
Clinicopathologic Features and Germline Sequence Variants in Young Patients (≤40 Years Old) With Pancreatic Ductal Adenocarcinoma.
CLINVAR
24366402 ↗
Summaries for patients. Assessing the genetic risk for BRCA-related breast or ovarian cancer in women: recommendations from the U.S. Preventive Services Task Force.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR