NM_000500.9:c.1439G>T (p.Arg480Leu) is a missense variant in exon 10 of CYP21A2, a gene associated with autosomal recessive congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The variant is present in gnomAD at an appreciable global allele frequency of 0.62% (v2.1, 1521/244182 alleles) and 0.33% (v4.1, 5128/1571116 alleles), exceeding the BS1 threshold (>0.3%) for a recessive disorder.1 In silico predictions are uniformly benign: REVEL score 0.334, BayesDel score -0.316 (predicting benign), and SpliceAI detects no splice alteration (max delta 0.00), satisfying BP4.2 ClinVar reports this variant as Benign by three clinical laboratories and Likely benign by three additional laboratories, with two VUS submissions, supporting BP6.3 Functional studies demonstrate the variant retains ~75-80% of wild-type 21-hydroxylase activity in transfected COS-1 cells, consistent with a mild functional impact, providing PS3 at the supporting level.4 Applying the generic ACMG/AMP 2015 combination rules (PMID:25741868), the criteria met are three supporting benign (BS1, BP4, BP6) and one supporting pathogenic (PS3). Likely benign requires at least two supporting benign criteria, which is satisfied.5
CYP21A2
Final classification
Likely Benign
CYP21A2 c.1439G>T · p.Arg480Leu
CYP21A2
NM_000500.9:c.1439G>T (p.Arg480Leu) is a missense variant in exon 10 of CYP21A2, a gene associated with autosomal recessive congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 supporting, BS1 supporting benign, BP4 supporting benign, BP6 supporting benign; combination = 1 supporting + 3 supporting benign, which maps to Likely Benign.
Classification rationale
PS3
BS1BP4BP6
Likely Benign
CYP21A2 c.1439G>T
PS3 + BS1 + BP4 + BP6
→
Likely Benign
2
revelbayesdelspliceai ↗
5
generic_acmg_combination_rules
Gene diagram
· NM_000500.9 · variants mapped to exon structure
CYP21A2
NM_000500.9
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 17 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
supporting
Pathogenic
Functional studies in transiently transfected COS-1 cells demonstrated that the p.Arg480Leu (R479L in older numbering) mutant retains approximately 75-80% of wild-type 21-hydroxylase activity toward both 17-hydroxyprogesterone and progesterone, consistent with a mild non-classical CAH phenotype. This partial reduction in enzyme activity supports a damaging effect at the supporting level.
PMID:17119906 (Robins et al. 2007): R479L showed 75.5% (SD 15.7%) activity for 17OHP and 79.6% (SD 12%) for progesterone vs WT in COS-1 cellskinetic analysis showed normal Km but mildly reduced Vmax
✓
BS1
supporting
Benign
BS1 requires allele frequency >0.3% in population databases for a recessive disorder. The variant is present in gnomAD v2.1 at global AF 0.62% (1521/244182 alleles) and in v4.1 at global AF 0.33% (5128/1571116 alleles). Both exceed the 0.3% threshold. Additionally, the variant was observed in heterozygous state in 1 of 92 healthy individuals in a Hungarian population study (PMID:19505723).
gnomAD v2.1 global AF: 0.62%gnomAD v4.1 global AF: 0.33%PMID:19505723: R479L found in 1/92 healthy Hungarian individuals
✓
BP4
supporting
Benign
BP4 requires multiple lines of computational evidence suggesting no impact. REVEL score is 0.334 (below 0.5 pathogenic threshold), BayesDel score is -0.316 (negative, predicts benign), and SpliceAI predicts no splice impact (max delta 0.00). All three in silico tools consistently predict a benign effect.
REVEL: 0.334 (benign range)BayesDel: -0.315808 (predicts benign)SpliceAI: max delta 0.00 (no splice impact)
✓
BP6
supporting
Benign
BP6 applies when a reputable source reports the variant as benign without access to the underlying evidence. ClinVar reports this variant as Benign by 3 clinical laboratories, Likely benign by 3 additional laboratories, and VUS by 2 laboratories. The majority clinical consensus across multiple submitters supports a benign interpretation.
ClinVar (VariationID 445854): Benign (3 labs)Likely benign (3 labs)VUS (2 labs)
Assessed · not applied
Pathogenic
PS1
PS1 requires a different amino acid change at the same residue that has been previously established as pathogenic.
PS2
No confirmed de novo occurrence with both parents confirmed unaffected was identified for this variant.
PS4
PS4 requires the prevalence of the variant in affected individuals to be significantly increased compared to controls.
PM1
PM1 requires location in a mutational hot spot or critical functional domain without benign variation.
PM2
PM2 requires the variant to be absent or at extremely low frequency in population databases (<0.1%).
PM6
PM6 requires an assumed de novo occurrence without confirmation of both parents.
PP1
PP1 requires cosegregation with disease in multiple affected family members.
PP2
PP2 requires a missense variant in a gene with a low rate of benign missense variation where missense variants are a common disease mechanism.
PP3
PP3 requires multiple lines of computational evidence supporting a deleterious effect.
PP4
PP4 requires the patient's phenotype or family history to be highly specific for a disease with a single genetic etiology.
PP5
PP5 requires a reputable source to have recently reported the variant as pathogenic.
Benign
BA1
BA1 requires allele frequency >1% in population databases.
BS2
BS2 requires observation in a healthy adult in a state (homozygous for recessive) where full penetrance is expected at an early age.
BS3
BS3 requires well-established functional studies showing no damaging effect.
BS4
BS4 requires lack of segregation in affected family members.
BP1
BP1 applies when a missense variant occurs in a gene where primarily truncating variants cause disease.
BP5
BP5 requires the variant to be found in a case with an alternate molecular basis for disease.
N/A · 4
PVS1 · PM5 · BP2 · BP7
Research & evidence
Population frequency · supports benign
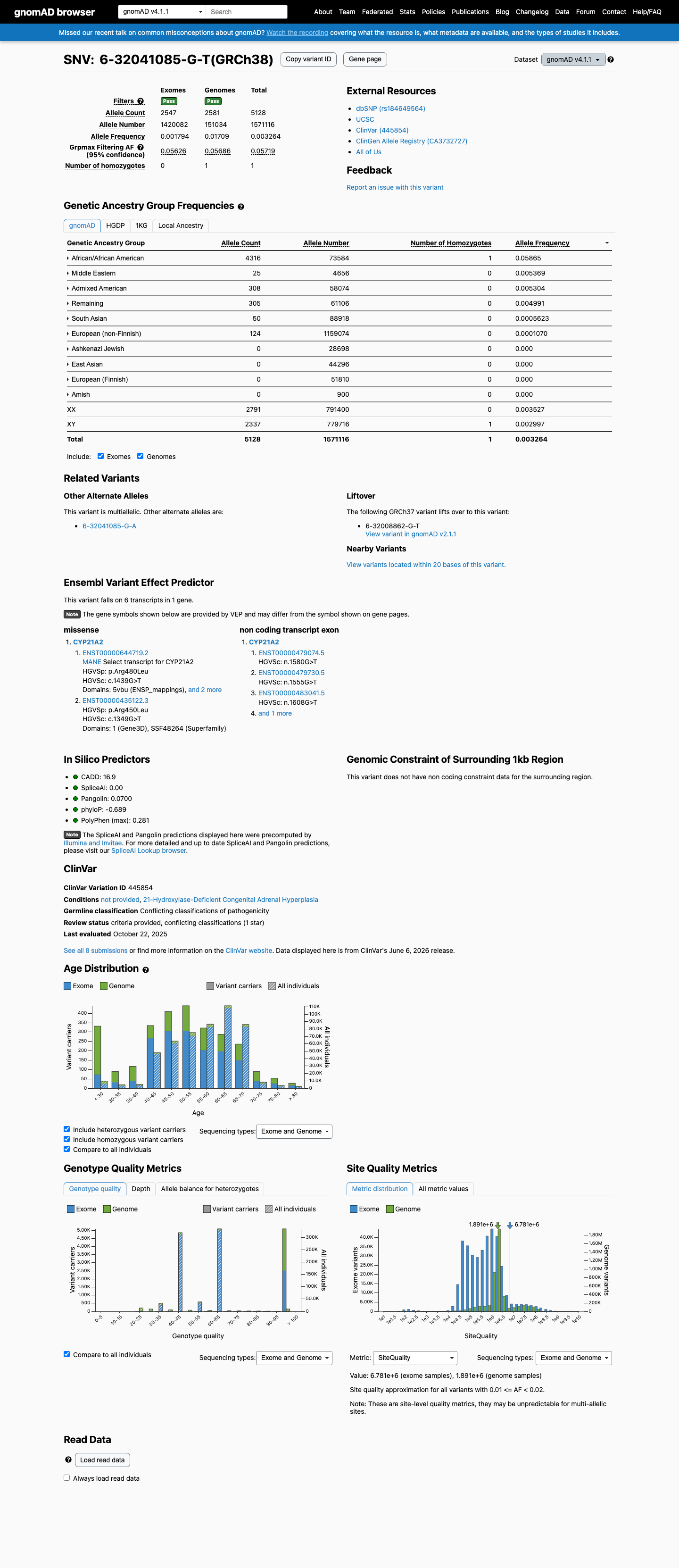
gnomAD v4.1
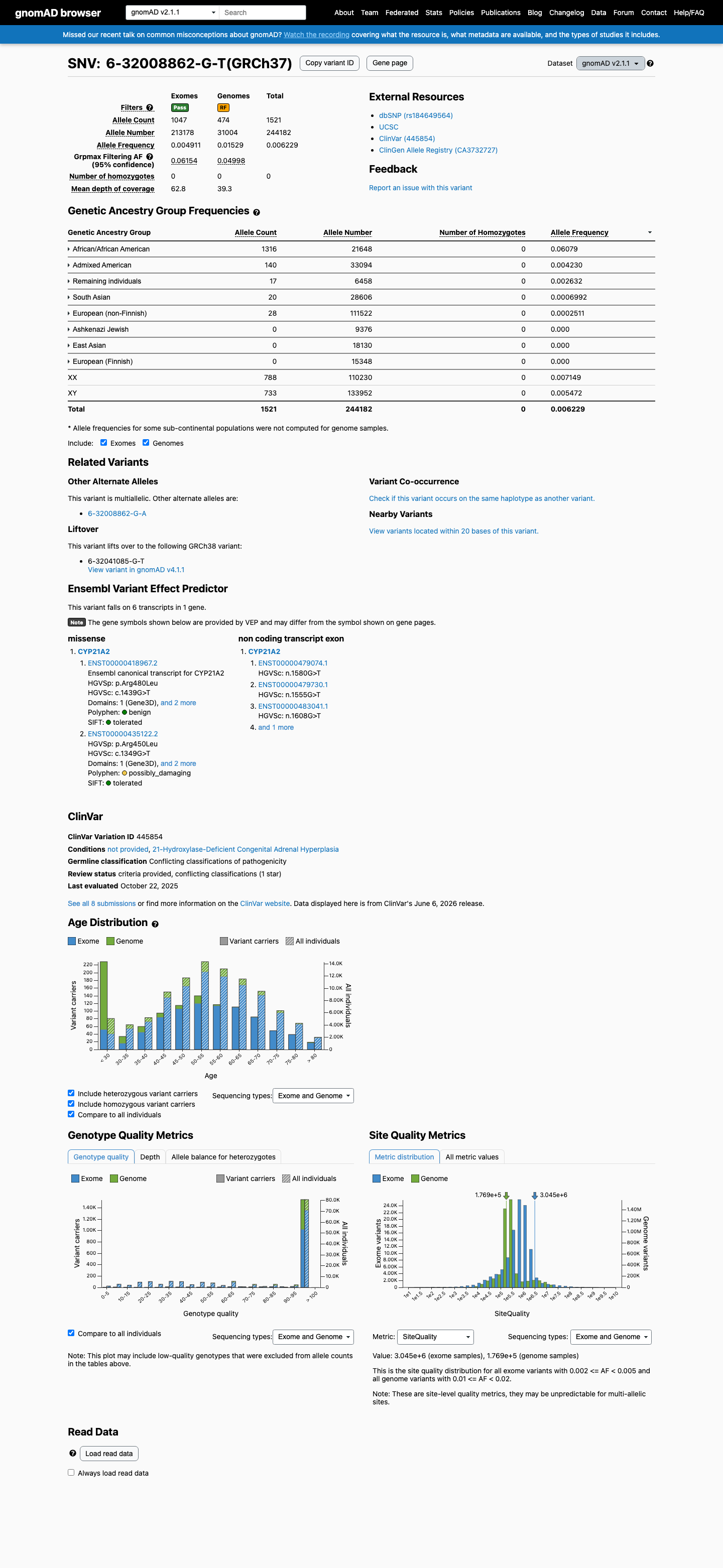
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00326392; MAF= 0.32639%, 5128/1571116 alleles, homozygotes = 1) and has highest observed frequency in the African/African American population (AF= 0.0586541; MAF= 5.86541%, 4316/73584 alleles, homozygotes = 1); grpmax FAF= 0.0571923.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00622896; MAF= 0.62290%, 1521/244182 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0.0607908; MAF= 6.07908%, 1316/21648 alleles, homozygotes = 0); grpmax FAF= 0.0615449.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.33%
· 5128 / 1,571,116
1 hom · FAF 5.7%
1 hom · FAF 5.7%
African/African American 4316 / 73,584 |
5.9% 1 hom |
Middle Eastern 25 / 4,656 |
0.54% |
Admixed American 308 / 58,074 |
0.53% |
Remaining individuals 305 / 61,106 |
0.5% |
South Asian 50 / 88,918 |
0.056% |
European (non-Finnish) 124 / 1,159,074 |
0.011% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.62%
· 1521 / 244,182
0 hom · FAF 6.2%
0 hom · FAF 6.2%
African/African American 1316 / 21,648 |
6.1% |
Admixed American 140 / 33,094 |
0.42% |
Remaining individuals 17 / 6,458 |
0.26% |
South Asian 20 / 28,606 |
0.07% |
European (non-Finnish) 28 / 111,522 |
0.025% |
+ 3 not observed (Ashkenazi Jewish, East Asian, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
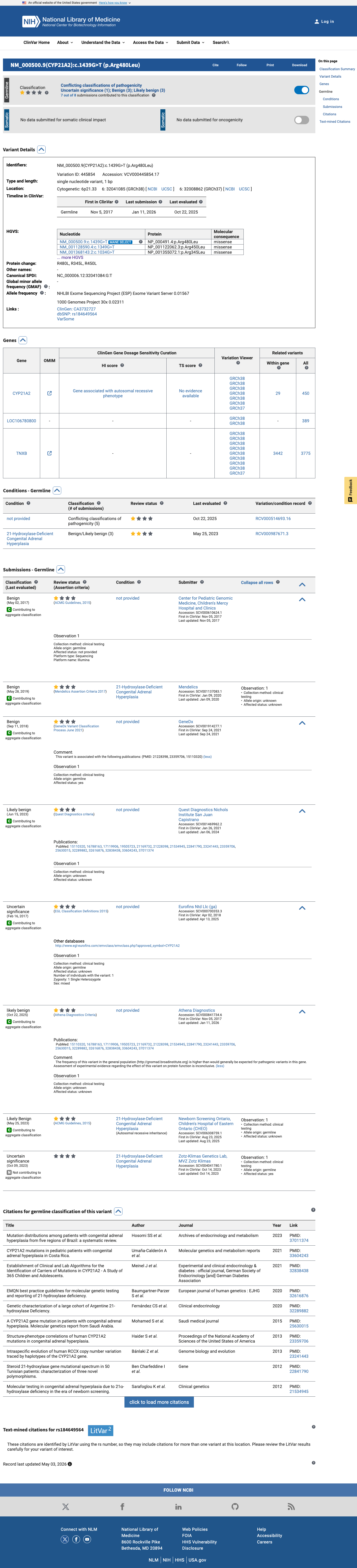
ClinVar
This variant has been reported in ClinVar as Benign (3 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Likely benign (1 clinical laboratory) and as likely benign (1 clinical laboratory) and as Likely Benign (1 clinical laboratory). (ClinVarID = 445854)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.334. BayesDel score = -0.315808.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
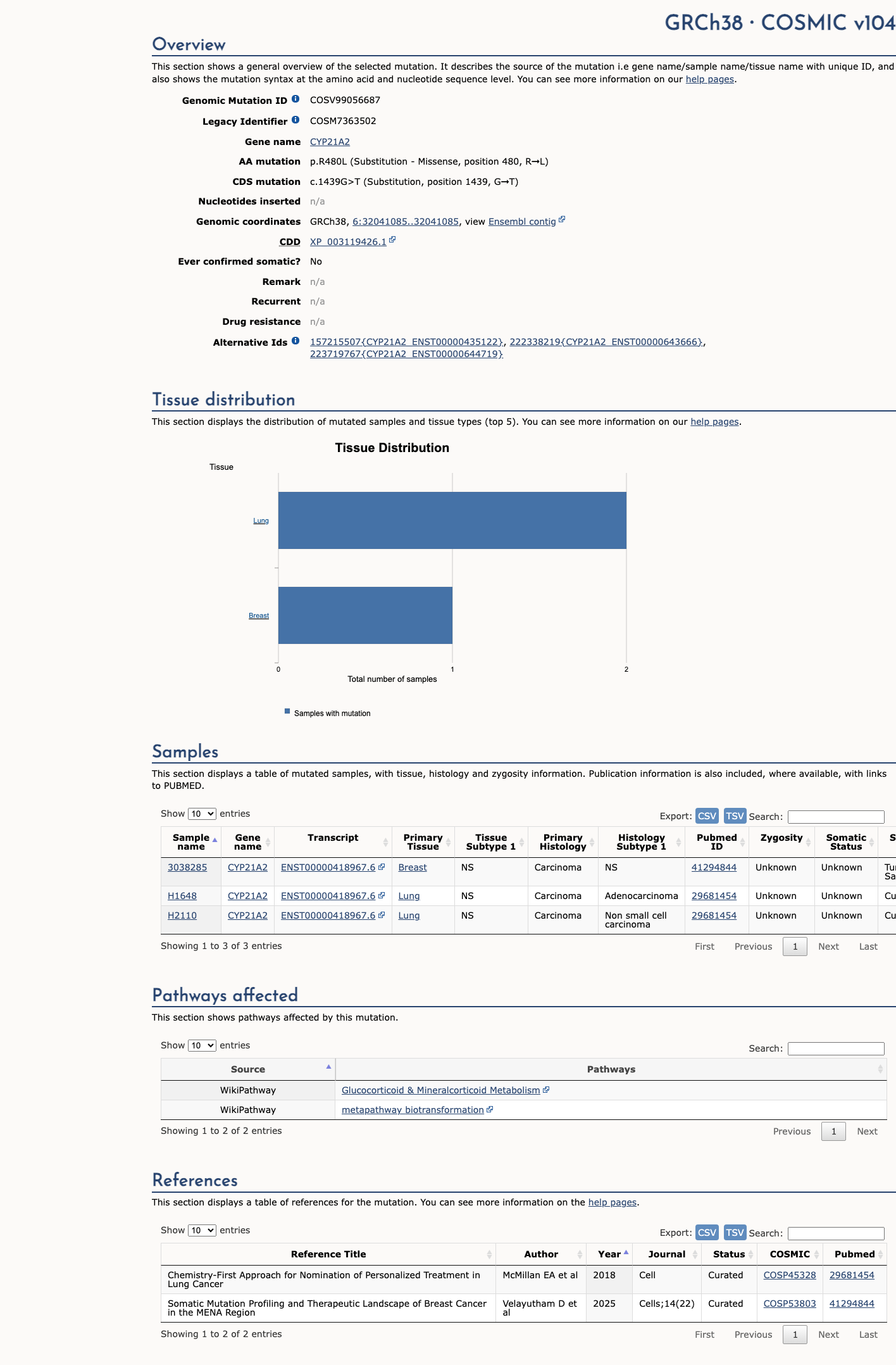
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV99056687, n = 3 times).
Hotspots
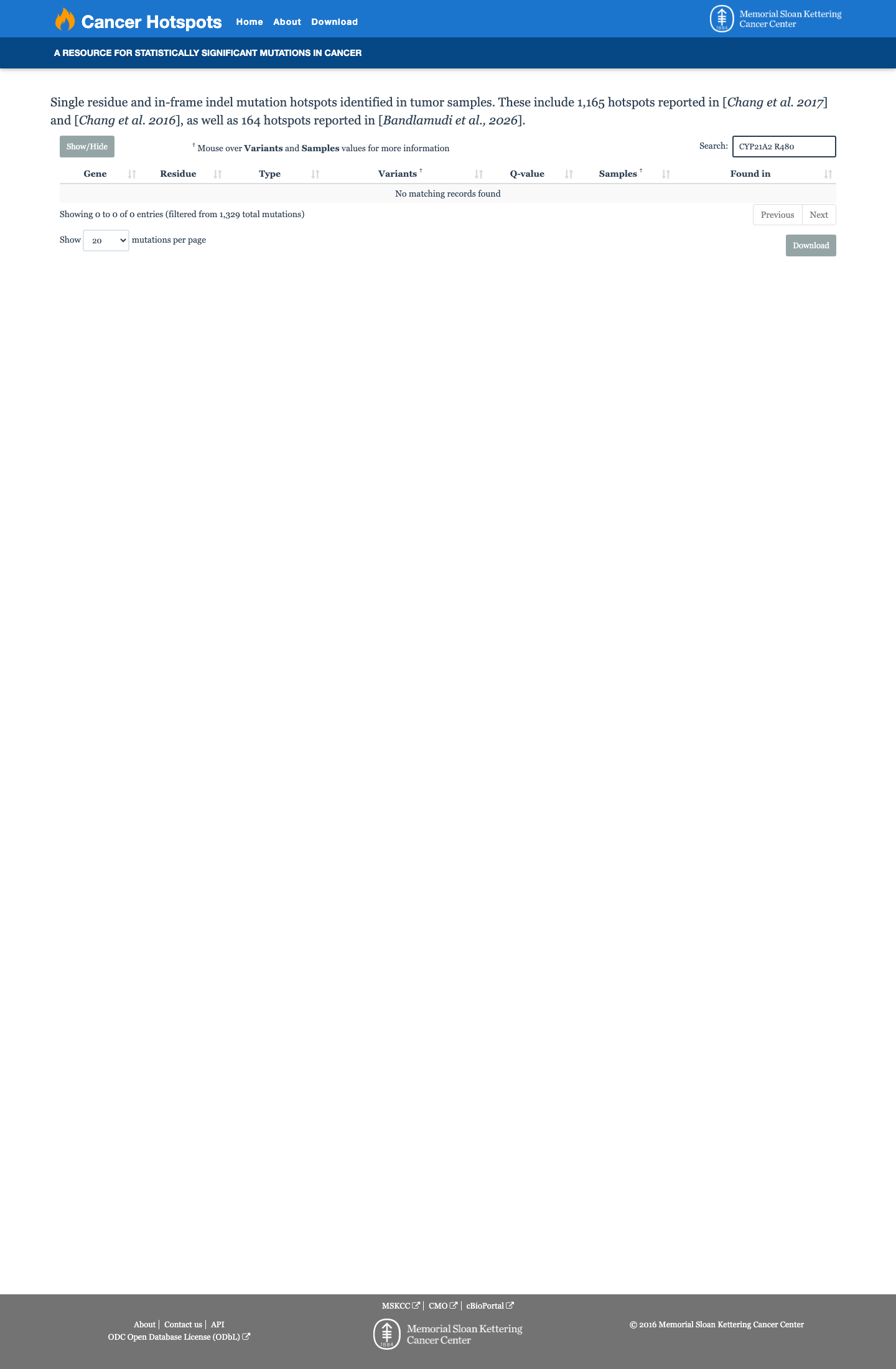
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Characterization of novel missense mutations in CYP21 causing congenital adrenal hyperplasia.
Found
(Robins et al.
Applied to
→PS3 supports · met
Linkage analysis of the C4A/C4B copy number variation and polymorphisms of the adjacent steroid 21-hydroxylase gene in a healthy population.
Found
gnomAD v2.1 global AF: 0.62% gnomAD v4.1 global AF: 0.33% PMID:19505723: R479L found in 1/92 healthy Hungarian individuals
Applied to
→BS1 supports · met
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
15110320 ↗
Detection and assignment of CYP21 mutations using peptide mass signature genotyping.
CLINVAR
16788163 ↗
Molecular model of human CYP21 based on mammalian CYP2C5: structural features correlate with clinical severity of mutations causing congenital adrenal hyperplasia.
CLINVAR
21169732 ↗
A large view of CYP21 locus among Sicilians and other populations: identification of a novel CYP21A2 variant in Sicily.
CLINVAR
21228398 ↗
Carrier testing for severe childhood recessive diseases by next-generation sequencing.
CLINVAR
23241443 ↗
Intraspecific evolution of human RCCX copy number variation traced by haplotypes of the CYP21A2 gene.
CLINVAR
23359706 ↗
Structure-phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia.
CLINVAR