c.1A>G (p.Met1Val) is a start-loss variant that abolishes the translation initiation codon of CYP21A2, a gene in which loss of function is a well-established mechanism for autosomal recessive congenital adrenal hyperplasia (21-hydroxylase deficiency).1 This variant is absent from gnomAD v2.1, v4.1, and gnomAD-Canada population databases.2 c.1A>G is classified as Pathogenic in ClinVar (Variation ID 1451630, submitted by Labcorp Genetics/Invitae) and independently reported as Pathogenic in a peer-reviewed study of CAH patients in the Anatolian population.3 Published evidence confirms that start codon mutations in CYP21A2 (including c.1A>G, M1I, and c.2T>C) result in complete loss of 21-hydroxylase enzymatic activity, with c.1A>G specifically associated with salt-wasting CAH.4 Applying generic ACMG/AMP 2015 combination rules: PVS1_VeryStrong (1) + PM2_Supporting (1) + PP5_Supporting (1) meets the threshold for Pathogenic classification (1 Very Strong + ≥2 Supporting).5
CYP21A2
Final classification
Pathogenic
CYP21A2 c.1A>G · p.Met1?
CYP21A2
c.1A>G (p.Met1Val) is a start-loss variant that abolishes the translation initiation codon of CYP21A2, a gene in which loss of function is a well-established mechanism for autosomal recessive congenital adrenal hyperplasia (21-hydroxylase deficiency).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 very strong, PM2 supporting, PP5 supporting; combination = 1 very strong + 2 supporting, which maps to Pathogenic.
Classification rationale
PVS1PM2PP5
Pathogenic
CYP21A2 c.1A>G
PVS1 + PM2 + PP5
→
Pathogenic
5
generic_acmg_combination_rules
Gene diagram
· NM_000500.9 · variants mapped to exon structure
CYP21A2
NM_000500.9
Fetching transcript structure from UCSC…
Applied criteria · 3 met · select any tile
Met
Not met
Not assessed
N/A
Strength
very strong
supporting
Pathogenic evidence
PVS
PS
PM
PP
Benign evidence
BA
BS
BP
—
—
—
Rationale
Select a criterion.
Sources
Evidence used
Gaps remaining
Rule
—
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
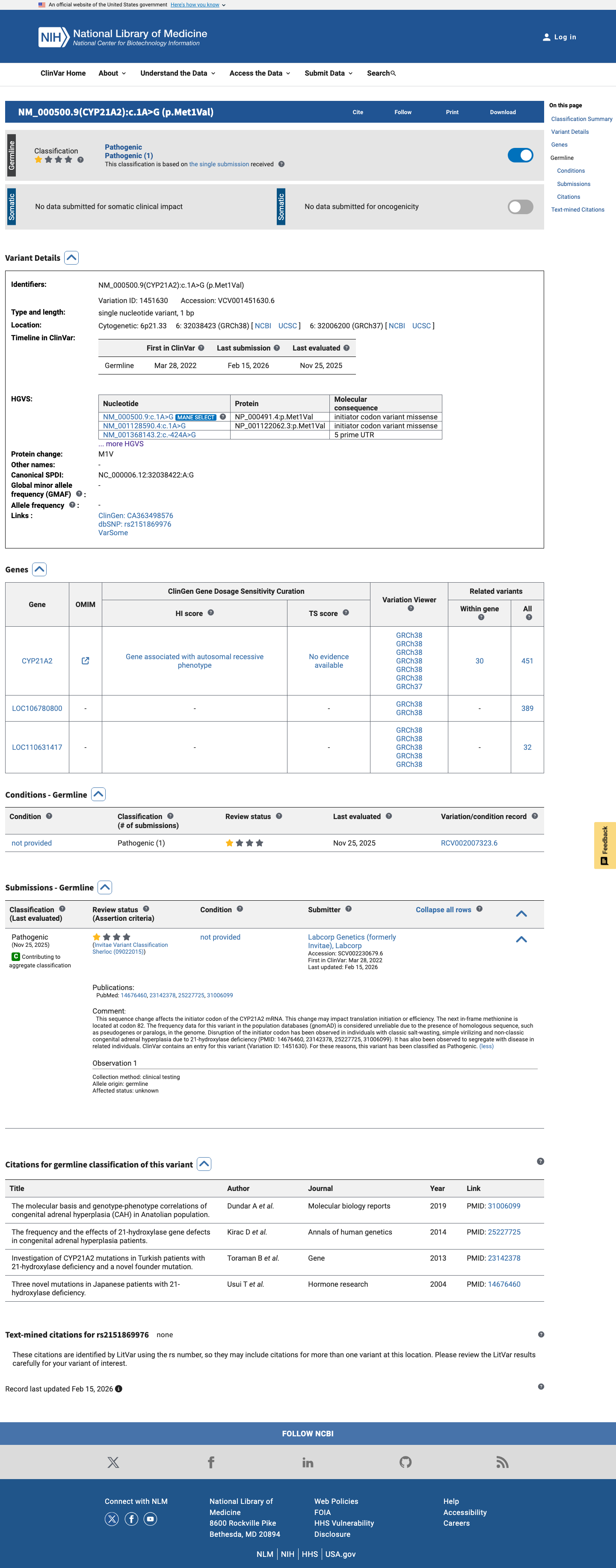
This variant has been reported in ClinVar as Pathogenic (1 clinical laboratory). (ClinVarID = 1451630)
In silico
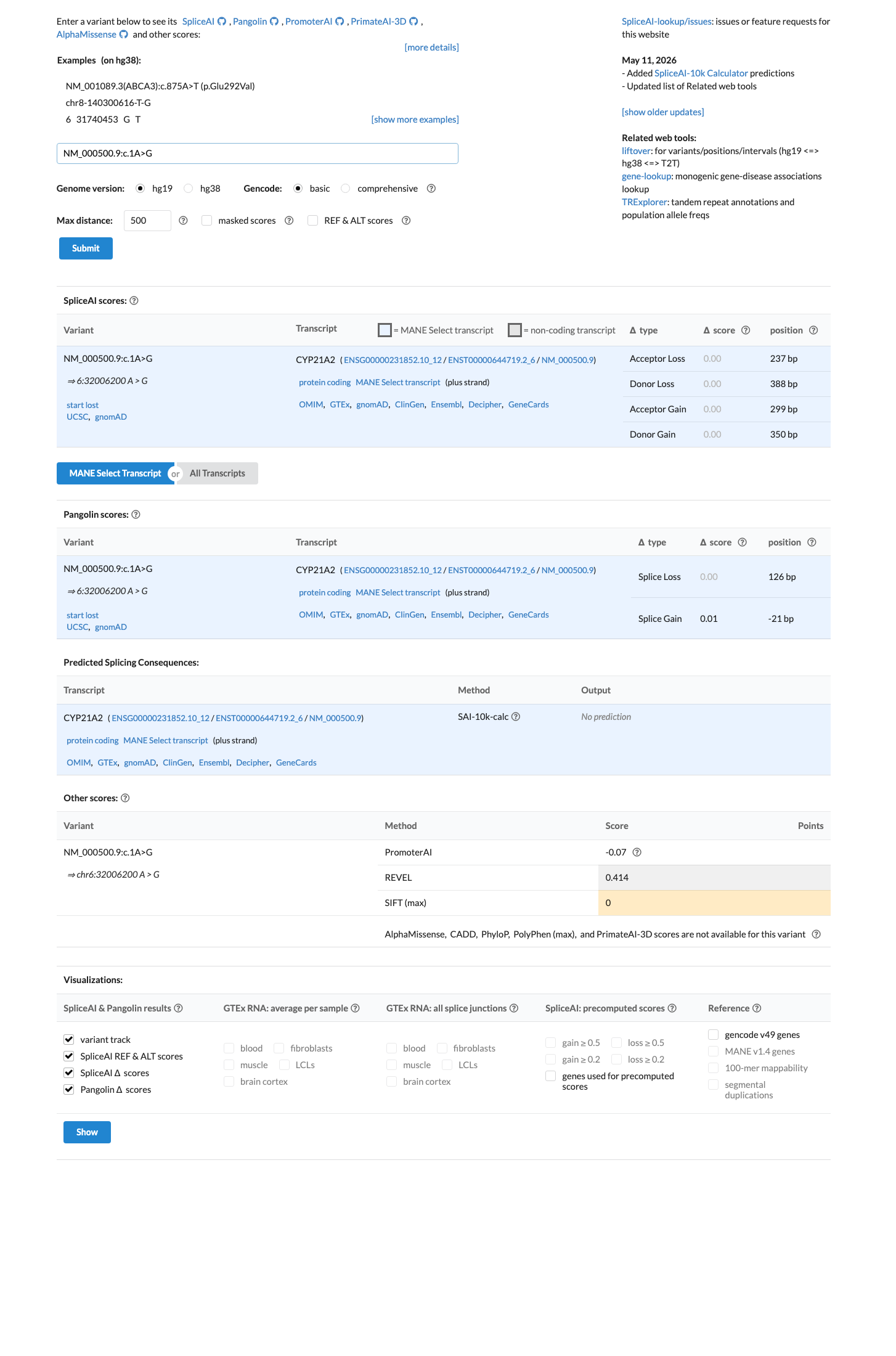
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.414. BayesDel score = 0.25807.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 3 further PMIDs triaged but not cited — see Sources & References.
Investigation of CYP21A2 mutations in Turkish patients with 21-hydroxylase deficiency and a novel founder mutation.
Searched
c.1A>GM1VM1?Met1Val
Found
Investigated CYP21A2 mutations in 48 Turkish CAH patients. Identified c.2T>C (p.M1?) as a novel founder mutation in 6 families (13.3% of mutant alleles), associated with classical CAH (salt-wasting in 5, simple virilizing in 1). In the discussion, c.1A>G is cited as being associated with salt-wasting CAH in an Iranian patient (Rabbani et al. 2012), supporting that disruption of the translation initiation codon causes severe disease.
Variant
✓ Names this variant — characterised directly
Applied to
→PVS1 supports · met
Why
Directly references c.1A>G as a pathogenic start codon variant associated with salt-wasting CAH. Cited for PVS1 assessment.
disruption of the first position of translation initiation codon, c.1A>G was shown to be associated with salt wasting form of 21OHD CAH in a patient from Iranian population as well (Rabbani et al., 2012)
Location Abstract; Results para 5-6; Table 1; Discussion para 3 (lines 623-625) · full text
The molecular basis and genotype-phenotype correlations of congenital adrenal hyperplasia (CAH) in Anatolian population.
Searched
c.1A>GM1Vp.M1Met1Val
Found
Comprehensive molecular analysis of CAH-associated genes (CYP21A2, CYP11B1, HSD3B2) in 365 Anatolian individuals. c.1A>G (p.M1V) was detected in heterozygous state in one CAH patient and classified as Pathogenic per ACMG guidelines. The variant was absent from population databases (ExAC, 1000 Genomes). The variant is identified as causing start loss and was among 9 CYP21A2 variants classified as Pathogenic in this study.
Variant
✓ Names this variant — characterised directly
Applied to
→PM2 supports · met
→PP5 supports · met
→PVS1 supports · met
Why
Directly reports c.1A>G as a pathogenic start-loss variant in a CAH patient. Cited for PVS1, PP5, and PM2 assessment.
p.M1 V [34, 35] / c.1A>G / Exon 1 / Missense, start loss ... Pathogenic / Pathogenic
Location Table 2, variant #16 (lines 501-510); Results section (lines 688-694) · full text
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
25227725 ↗
The frequency and the effects of 21-hydroxylase gene defects in congenital adrenal hyperplasia patients.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR