NM_000516.5:c.602G>A (p.Arg201His) is a well-characterized activating missense variant in the GNAS gene. Functional studies demonstrate that this substitution reduces GTPase activity approximately 30-fold, causing constitutive activation of adenylyl cyclase and downstream cAMP signaling (PS3_Strong).1 The variant resides at codon 201 within the GTPase domain of Gsα, a critical functional domain and established mutational hotspot (PM1_Moderate).2 A different pathogenic missense change at the same residue, p.Arg201Cys, is well-established in McCune-Albright syndrome and functionally equivalent (PM5_Moderate).3 The variant has been reported in numerous individuals with McCune-Albright syndrome; Lumbroso et al. identified R201H in 34 of 113 patients with MAS features (PS4_Moderate).4 The variant is extremely rare in population databases (gnomAD v2.1 AF=0.00159%), supporting pathogenicity (PM2_Supporting).5 Computational predictors strongly support a deleterious effect: REVEL score 0.969, BayesDel score 0.612 (PP3_Supporting).6 ClinVar classifies this variant as Pathogenic (Variation ID 15934) with consensus across multiple clinical laboratories (PP5_Supporting).7 Applying the generic ACMG/AMP 2015 combination rules: one Strong criterion (PS3) plus three Moderate criteria (PM1, PM5, PS4) and three Supporting criteria (PM2, PP3, PP5) meets the threshold for Pathogenic classification.8
GNAS
Final classification
Pathogenic
GNAS c.602G>A · p.Arg201His
GNAS
NM_000516.5:c.602G>A (p.Arg201His) is a well-characterized activating missense variant in the GNAS gene. Functional studies demonstrate that this substitution reduces GTPase activity approximately 30-fold, causing constitutive activation of adenylyl cyclase and downstream cAMP signaling (PS3_Strong).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 strong, PS4 moderate, PM1 moderate, PM2 supporting, PM5 moderate, PP3 supporting, PP5 supporting; combination = 1 strong + 3 moderate + 3 supporting, which maps to Pathogenic.
Classification rationale
PS3PS4PM1PM2PM5PP3PP5
Pathogenic
GNAS c.602G>A
PS3 + PS4 + PM1 + PM2 + PM5 + PP3 + PP5
→
Pathogenic
6
revelbayesdel
8
generic_acmg_combination_rules
Gene diagram
· NM_000516.5 · variants mapped to exon structure
GNAS
NM_000516.5
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 17 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
strong
Pathogenic
Well-established functional studies demonstrate that p.Arg201His constitutively activates Gsα. Landis et al. (PMID:2549426) showed that R201H substitution reduces GTPase activity approximately 30-fold, resulting in constitutive adenylyl cyclase activation. Taki et al. (PMID:26257060) demonstrated that transgenic expression of GNAS R201H in murine pancreas elevates cAMP levels, induces ductal dilation, and cooperates with KrasG12D to promote tumorigenesis recapitulating human IPMN. Two independent publications with direct variant-specific functional characterization satisfy PS3 at strong strength.
In vitro mutagenesis: R201H reduces GTPase activity ~30-fold with constitutive adenylyl cyclase activation (PMID:2549426).In vivo mouse model: GNAS R201H expression elevates cAMP and promotes pancreatic tumorigenesis (PMID:26257060).OncoKB classification: Oncogenic
✓
PS4
moderate
Pathogenic
This variant has been reported in numerous unrelated individuals with McCune-Albright syndrome and related phenotypes. Lumbroso et al. (PMID:15126527) identified R201H in 34 of 113 patients with MAS signs, representing the most common activating mutation. The variant is classified as Pathogenic in ClinVar by multiple clinical laboratories (Variation ID 15934). gnomAD population frequency is extremely low (AF=0.00159%; 4/251,454 alleles in v2.1), demonstrating significant enrichment in affected individuals compared to population controls. Strength is calibrated to moderate given the predominantly somatic mosaic context.
34/113 patients with MAS signs carry R201H (PMID:15126527).Multiple independent case reports and series document R201H in MAS.ClinVar: 6 labs classify as Pathogenic
✓
PM1
moderate
Pathogenic
The variant resides at codon 201 within the GTPase domain of Gsα, a critical functional domain. Arg201 is the site of ADP-ribosylation by cholera toxin and is essential for intrinsic GTPase activity. The codon is a well-established mutational hotspot in McCune-Albright syndrome, pituitary adenomas, and other tumors. Cancerhotspots.org identifies this residue as statistically significant. The variant lies within a domain where essentially all reported missense changes are pathogenic.
Codon 201 is in the GTPase domain of Gsαcritical for GTP hydrolysis.Arg201 is the site of cholera toxin ADP-ribosylation that inactivates GTPase.
✓
PM2
supporting
Pathogenic
This variant is present at extremely low frequency in population databases. gnomAD v2.1 exomes: allele frequency 1.59×10⁻⁵ (0.00159%; 4/251,454 alleles, 0 homozygotes). gnomAD v4.1: allele frequency 2.48×10⁻⁵ (40/1,613,098 alleles, 0 homozygotes). Both are well below the 0.1% threshold for PM2. The variant is absent from gnomAD-Canada v1.0.
gnomAD v2.1: AF=1.59e-05 (4/251454 alleles).gnomAD v4.1: AF=2.48e-05 (40/1
✓
PM5
moderate
Pathogenic
A different missense change at the same amino acid residue (p.Arg201Cys, resulting from c.601C>T) is well-established as pathogenic. R201C is the second most common activating GNAS mutation in McCune-Albright syndrome and related disorders, with equivalent gain-of-function consequences. Lumbroso et al. (PMID:15126527) reported R201C in 15/113 patients. Both R201H and R201C are established activating mutations that disrupt GTPase activity and constitutively activate adenylyl cyclase.
R201C (c.601C>T) is a well-established pathogenic missense at the same codon.PMID:15126527: R201C found in 15/49 mutation-positive MAS patients.PMID:2549426: R201C also characterized as activating with GTPase inhibition.
✓
PP3
supporting
Pathogenic
Multiple lines of computational evidence support a deleterious effect. REVEL score is 0.969, which is strongly predictive of pathogenicity. BayesDel score is 0.612, consistent with a damaging prediction. SpliceAI predicts no significant splice impact (max delta = 0.01), which is expected for a missense variant not near splice junctions. The REVEL score in particular provides strong in silico support.
REVEL score: 0.969 (strongly pathogenic).BayesDel score: 0.612 (damaging).SpliceAI max delta: 0.01 (no predicted splice impact).
✓
PP5
supporting
Pathogenic
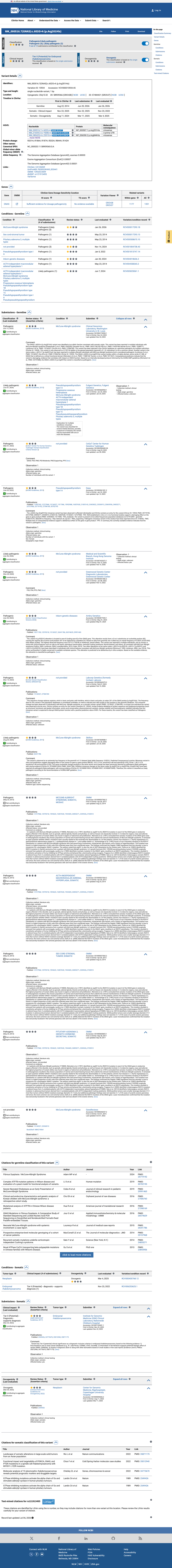
This variant is reported as Pathogenic in ClinVar (Variation ID 15934) by multiple clinical laboratories: 6 classify as Pathogenic and 3 as Likely pathogenic. While the aggregate review status is 'criteria provided, single submitter' (no expert panel review), the consensus across multiple independent laboratories provides supporting evidence for pathogenicity.
ClinVar Variation ID 15934: classified as Pathogenic.6 clinical laboratories report Pathogenic3 report Likely pathogenic.
Assessed · not applied
Pathogenic
PS1
PS1 requires the same amino acid change (p.Arg201His) resulting from a different nucleotide change.
PS2
No confirmed de novo occurrence with both maternity and paternity confirmed has been identified for this variant in a germline context.
PM6
PM6 applies when a variant is assumed de novo but without confirmation of paternity and maternity.
PP1
No cosegregation data are available for this variant in a germline context.
PP2
PP2 requires that the gene has a low rate of benign missense variation and that missense variants are a common mechanism of disease.
PP4
PP4 requires that the patient's phenotype or family history is highly specific for a disease with a single genetic etiology.
Benign
BA1
BA1 requires allele frequency >1% in population databases.
BS1
BS1 requires allele frequency >0.3% in population databases.
BS2
BS2 requires observation in healthy adults at significant frequency.
BS3
BS3 requires well-established functional studies showing no damaging effect.
BS4
BS4 requires lack of segregation in affected family members.
BP1
BP1 applies when a missense variant occurs in a gene where only truncating variants cause disease.
BP2
BP2 requires observation in trans with a pathogenic variant in a gene associated with a recessive disorder.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on gene or gene product.
BP5
BP5 requires that the variant is found in a case with an alternate molecular basis for disease.
BP6
BP6 requires a reputable source to report the variant as benign.
BP7
BP7 applies to synonymous variants with no predicted splice impact.
N/A · 1
PVS1
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
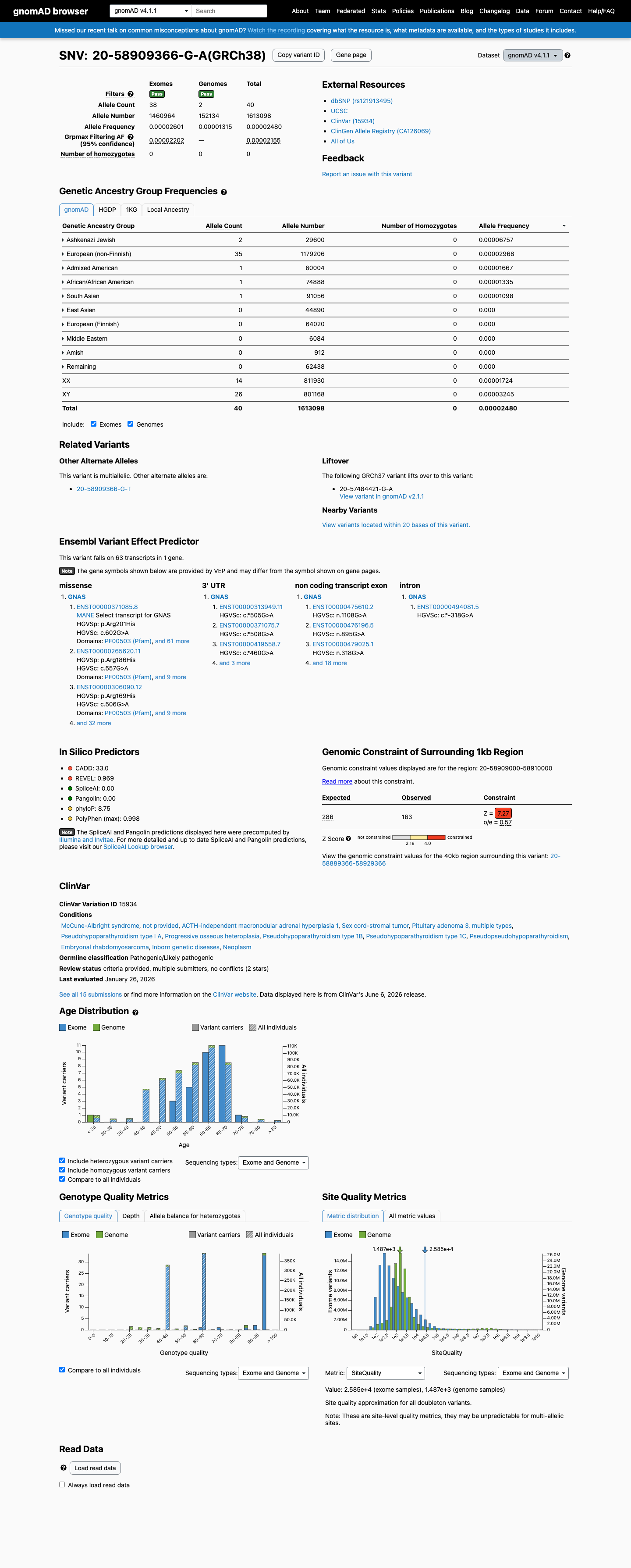
This variant is present in gnomAD v4.1 (AF= 2.4797e-05; MAF= 0.00248%, 40/1613098 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 6.75676e-05; MAF= 0.00676%, 2/29600 alleles, homozygotes = 0); grpmax FAF= 2.155e-05.
v2.1
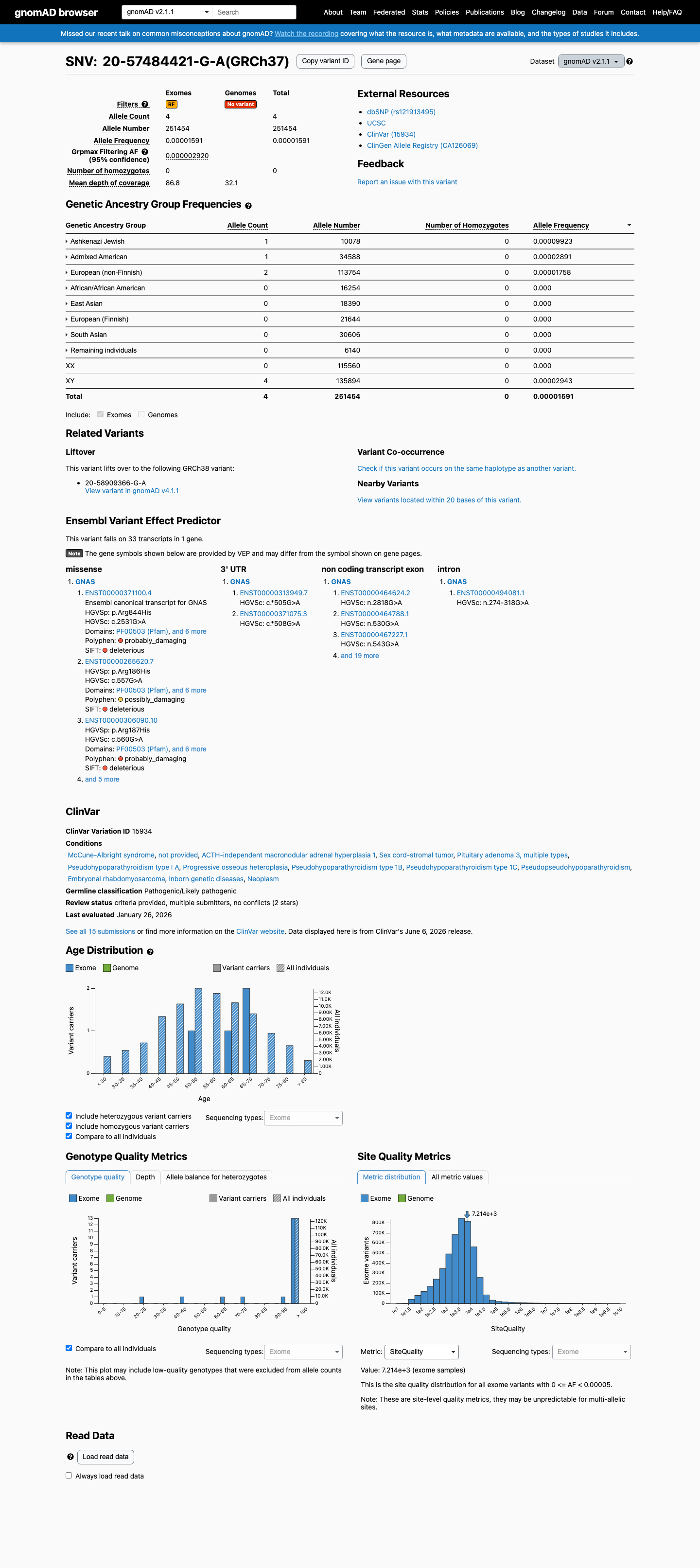
This variant is present in gnomAD v2.1 (AF= 1.59075e-05; MAF= 0.00159%, 4/251454 alleles, homozygotes = 0) and has highest observed frequency in the Ashkenazi Jewish population (AF= 9.9226e-05; MAF= 0.00992%, 1/10078 alleles, homozygotes = 0); grpmax FAF= 2.92e-06.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00010860121633362293, 2/18416 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0025%
· 40 / 1,613,098
0 hom · FAF 0.0022%
0 hom · FAF 0.0022%
Ashkenazi Jewish 2 / 29,600 |
0.0068% |
European (non-Finnish) 35 / 1,179,206 |
0.003% |
Admixed American 1 / 60,004 |
0.0017% |
African/African American 1 / 74,888 |
0.0013% |
South Asian 1 / 91,056 |
0.0011% |
+ 5 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern)
gnomAD v2.1
0.0016%
· 4 / 251,454
0 hom · FAF 0.00029%
0 hom · FAF 0.00029%
Ashkenazi Jewish 1 / 10,078 |
0.0099% |
Admixed American 1 / 34,588 |
0.0029% |
European (non-Finnish) 2 / 113,754 |
0.0018% |
+ 5 not observed (African/African American, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
0.011%
· 2 / 18,416
0 hom · FAF 0.003%
0 hom · FAF 0.003%
European (non-Finnish) 2 / 11,738 |
0.017% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
ClinVar
This variant has been reported in ClinVar as Pathogenic (6 clinical laboratories) and as Likely pathogenic (3 clinical laboratories). (ClinVarID = 15934)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.969. BayesDel score = 0.611857.
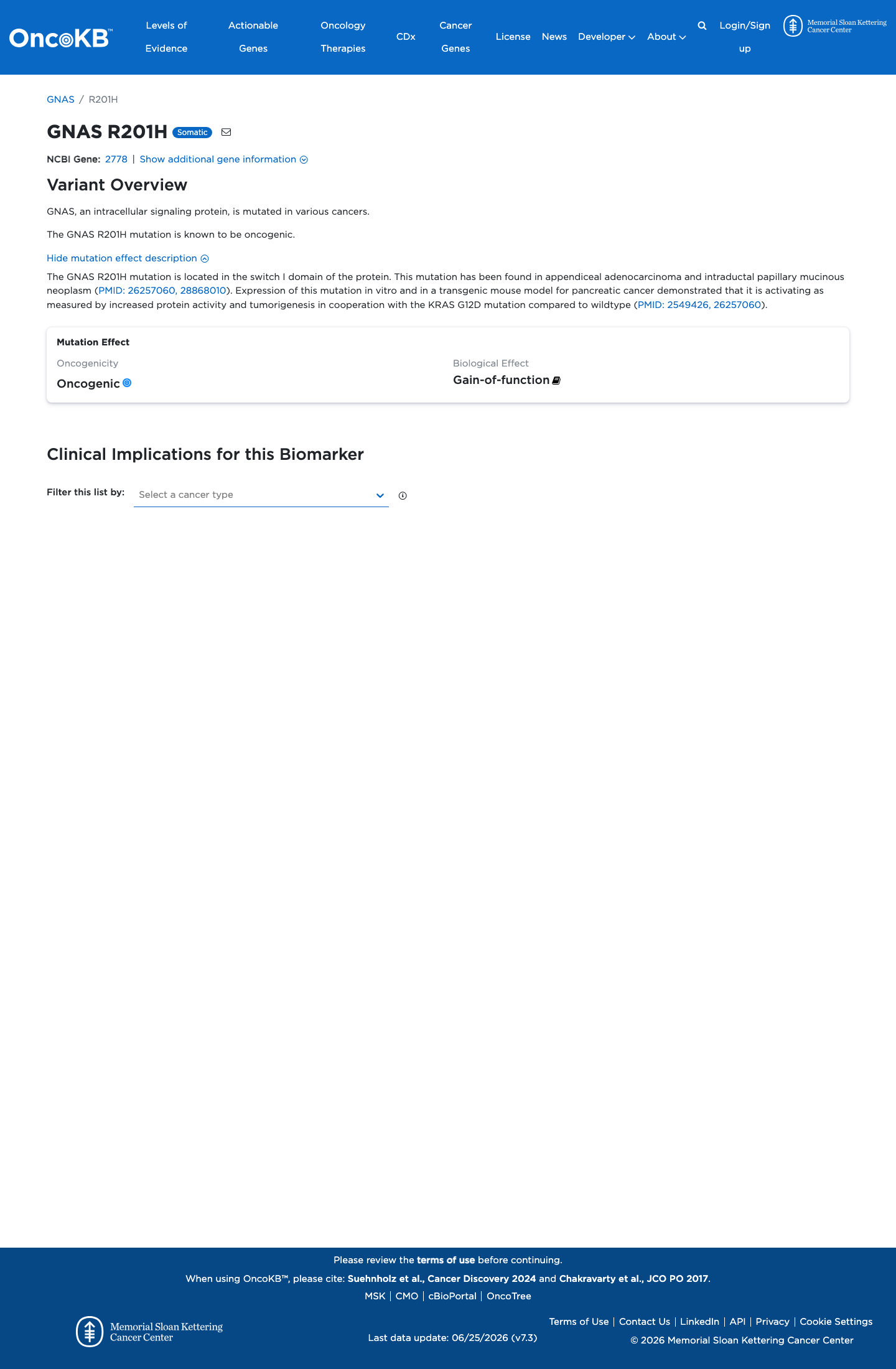
Functional
Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Gain-of-function; curated oncogenicity label: Oncogenic.

COSMIC
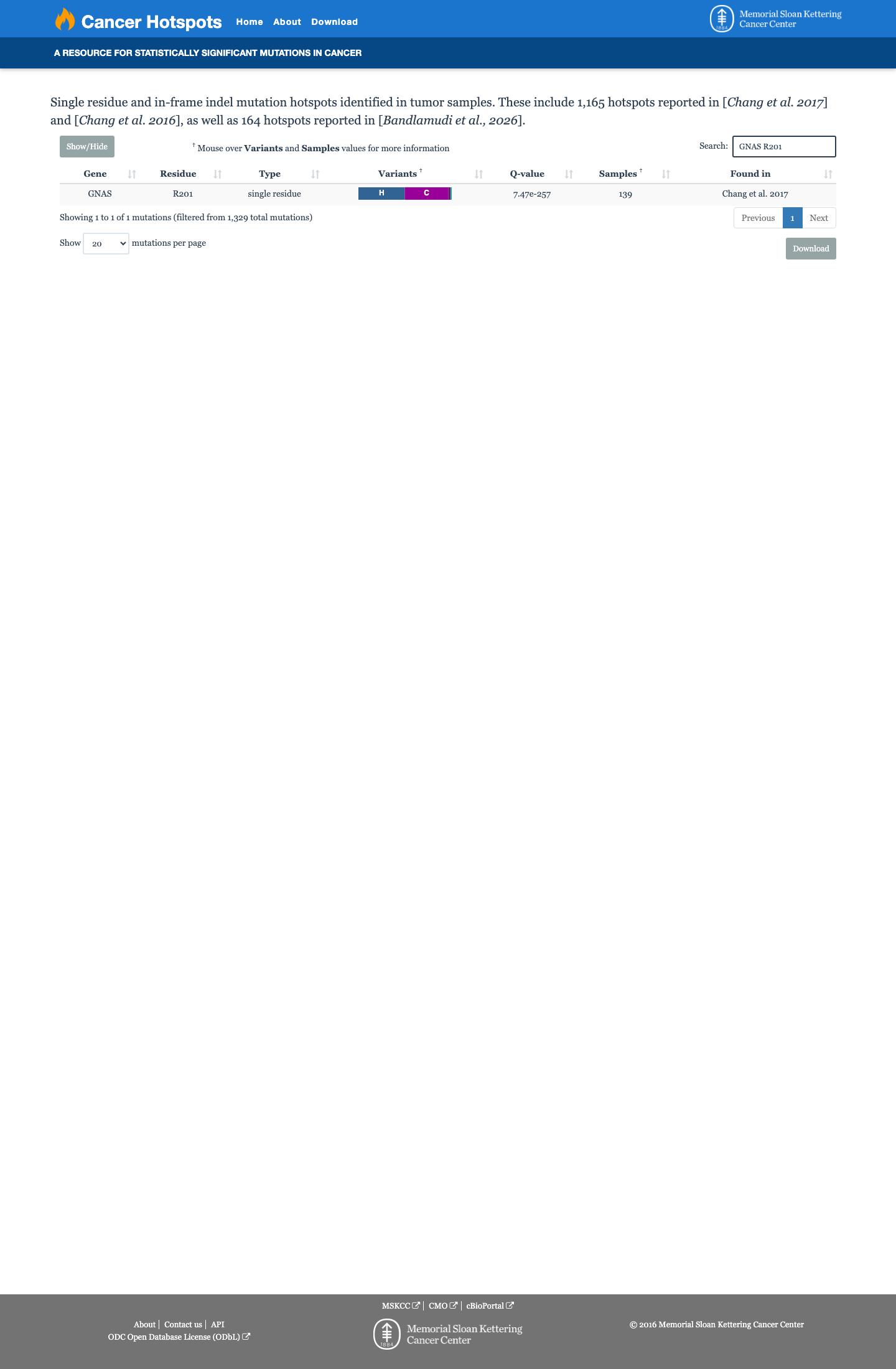
Cancer hotspots
Somatic evidence
Hotspot
COSMIC
This variant lies in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
4papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 4 further PMIDs triaged but not cited — see Sources & References.
Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study.
Searched
c.602G>Ap.Arg201HisR201HhistidineArg201
Found
Lumbroso et al. conducted a systematic search for Gsα activating mutations in 113 patients with signs of McCune-Albright syndrome. The Arg201His mutation was identified in 34 of 49 mutation-positive patients (69%), making it the most common activating mutation. The mutation was detected in multiple tissue types including ovary, bone, liver, and blood.
Variant
✓ Names this variant — characterised directly
Applied to
→PM5 supports · met
→PS4 supports · met
Why
Variant-specific prevalence data confirmed; 34/113 patients with R201H. Cited for PS4 (multiple probands) and PM5 (R201C as pathogenic comparator at same residue).
A mutation of arginine 201 in the Gsα protein was found in 49 of the 113 patients (43%), with a net preponderance of the substitution by histidine (n = 34) as opposed to cysteine (n = 15).
Location Abstract; Results para 1-4; Table 5 · Context PCR-based enrichment method with EagI restriction digestion on DNA from 174 tissue samples across 113 patients · full text
Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome.
Searched
c.602G>Ap.Arg201HisR201HArg201Arg201His
Found
Schwindinger et al. identified the c.602G>A (p.Arg201His) mutation in a 53-year-old man with McCune-Albright syndrome. The mutation was detected in peripheral blood leukocytes and at low levels in skin, confirming somatic mosaicism. The authors discuss prior in vitro mutagenesis data showing that Arg201 substitutions reduce GTPase activity ~30-fold and constitutively activate adenylyl cyclase.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS4 supports · met
Why
Variant-specific identification confirmed; first report of R201H in MAS patient tissue. Cited for PS4 (case observation) and PM1 (functional domain context).
This single-base substitution results in the replacement of arginine by histidine at position 201 of the mature Gsα protein.
Location Abstract; Results para 1-3; Discussion para 1-4 · Context Denaturing gradient gel electrophoresis, allele-specific PCR, single-nucleotide primer extension on leukocyte and skin DNA from a single MAS patient · full text
GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours.
Searched
Arg201R201HR201CgspGNAS
Found
Landis et al. identified somatic mutations in the GNAS gene (then termed gsp oncogene) in a subset of human growth hormone-secreting pituitary tumors. The mutations replaced Arg201 with either Cys or His and Gln227 with Arg, resulting in inhibition of GTPase activity and constitutive activation of adenylyl cyclase. In vitro mutagenesis demonstrated that Arg201 substitutions reduce GTPase activity approximately 30-fold.
Variant
✓ Names this variant
Applied to
→PS3 supports · met
Why
Variant-specific functional data confirmed; original characterization of R201H as an activating mutation. Cited for PS3 at strong level (landmark functional study).
A subset of growth hormone-secreting human pituitary tumours carries somatic mutations that inhibit GTPase activity of a G protein alpha chain, alpha(s). The resulting activation of adenylyl cyclase bypasses the cells' normal requirement for trophic hormone.
Location Abstract · Context In vitro mutagenesis and GTPase activity assays on Gsα; functional characterization in S49 cyc- murine lymphoma cells (as cited by PMID:1594625)
GNAS(R201H) and Kras(G12D) cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm.
Searched
c.602G>AR201HGNASR201HArg201His
Found
Taki et al. generated transgenic mice conditionally expressing GNAS R201H in the pancreas. GNAS R201H expression alone caused elevated cAMP levels, pancreatic ductal dilation, loss of acinar cells, and fibrosis. When combined with Kras G12D, GNAS R201H cooperatively promoted macroscopic cystic pancreatic tumors recapitulating human intraductal papillary mucinous neoplasm within 5 weeks.
Variant
✓ Names this variant — characterised directly
Applied to
→PS3 supports · met
Why
Variant-specific functional data confirmed in in vivo model; demonstrates constitutive Gsα activation and oncogenic cooperation. Cited for PS3 at strong level alongside PMID:2549426.
The GNASR201H mutation results in constitutive activation of Gsα.
Location Abstract; Results and Discussion para 1-6; Figure 1-4 · Context Transgenic mouse model (Tg(CAG-LSL-GNASR201H);Ptf1aCre/+); cAMP measurement, histopathology, Kaplan-Meier survival analysis · full text
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
28868010 ↗
Clinical Benefit from Trametinib in a Patient with Appendiceal Adenocarcinoma with a GNAS R201H Mutation.
ONCOKB
24855271 ↗
Recurrent somatic mutations underlie corticotropin-independent Cushing's syndrome.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR