NM_000535.7:c.2566C>T (p.Leu856=) is a synonymous variant in exon 15 of PMS2. It is extremely rare in population databases, with an allele frequency of 5.057e-06 in gnomAD v4.1 (5/988,724 alleles, no homozygotes) and absent from gnomAD v2.1, satisfying PM2_Supporting per PMS2 VCEP v2.0.0.1 SpliceAI predicts no splicing impact for this variant (max delta = 0.00), satisfying BP4_Supporting per PMS2 VCEP v2.0.0 for synonymous variants.2 The variant is a synonymous substitution located at c.2566 in exon 15, +120 nucleotides from the splice acceptor site, well beyond the +7 position defined by the BP7 rule. BP7_Supporting is met per PMS2 VCEP v2.0.0.3 PVS1, PS1, and PM5 are not applicable as this is a synonymous variant producing no amino acid change. PS4, PM1, PM6, PP2, PP5, BP1, BP2, and BP6 are not applicable per the PMS2 VCEP v2.0.0 specifications.4 No de novo observations (PS2), functional studies (PS3, BS3), cosegregation data (PP1, BS4), trans co-occurrence data (BS2), tumor pathology data (PP4, BP5), or computational evidence supporting pathogenicity (PP3) were identified for this variant. This variant has been reported in ClinVar as Likely benign by four clinical laboratories and Benign by one clinical laboratory (ClinVar ID 701564). In ClinVar, submissions cited methodology papers without variant-specific clinical evidence.5 Under the PMS2 VCEP v2.0.0 combination rules, the presence of two benign supporting criteria (BP4_Supporting, BP7_Supporting) satisfies Rule19 (>=2 Benign.Supporting -> Likely Benign). PM2_Supporting is also met but does not alter the classification under the VCEP rule hierarchy.6
PMS2
Final classification
Likely Benign
PMS2 c.2566C>T · p.Leu856=
PMS2
NM_000535.7:c.2566C>T (p.Leu856=) is a synonymous variant in exon 15 of PMS2. It is extremely rare in population databases, with an allele frequency of 5.057e-06 in gnomAD v4.1 (5/988,724 alleles, no homozygotes) and absent from gnomAD v2.1, satisfying PM2_Supporting per PMS2 VCEP v2.0.0.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PMS2 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule19 (Benign.Supporting >=2) with applied criteria: PM2 supporting, BP4 supporting benign, BP7 supporting benign; maps to Likely Benign.
Classification rationale
PM2
BP4BP7
Likely Benign
PMS2 c.2566C>T
PM2 + BP4 + BP7
→
Likely Benign
Gene diagram
· NM_000535.7 · variants mapped to exon structure
PMS2
NM_000535.7
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 11 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000535.7:c.2566C>T is extremely rare in population databases. The variant is absent from gnomAD v2.1 and present in gnomAD v4.1 at an allele frequency of 5.057e-06 (5/988,724 alleles, no homozygotes), which is below the PMS2 VCEP v2.0.0 PM2_Supporting threshold of <0.00002 (<1 in 50,000 alleles). The grpmax filtering allele frequency is 1.39e-06.
gnomAD v4.1: AF=5.057e-06 (5/988724 alleles0 homozygotes)
✓
BP4
supporting
Benign
NM_000535.7:c.2566C>T is a synonymous variant and SpliceAI predicts no splicing impact (max delta score = 0.00), which satisfies the PMS2 VCEP v2.0.0 BP4_Supporting rule for intronic and synonymous variants (SpliceAI delta score <= 0.1). The HCI prior branch of BP4 does not apply as this is not a missense variant.
SpliceAI max delta = 0.00confirming no predicted splicing impact. Synonymous variant meets VCEP BP4_Supporting threshold.
✓
BP7
supporting
Benign
NM_000535.7:c.2566C>T is a synonymous (silent) variant located in exon 15 of PMS2 (c.2446-c.*2474) at position c.2566, which is +120 nucleotides downstream of the splice acceptor site. This is beyond the +7 position from the 3' exon boundary, satisfying the PMS2 VCEP v2.0.0 BP7 rule for synonymous variants at or beyond -21/+7 (5'/3' exonic). The variant does not lie within the critical splice consensus region.
Synonymous variant at c.2566 in exon 15located +120 from splice acceptor (beyond +7 threshold). SpliceAI max delta = 0.00 confirms no cryptic splice effect.
Assessed · not applied
Pathogenic
PS2
No de novo occurrences of NM_000535.7:c.2566C>T were identified in the literature or curated de novo databases.
PS3
No well-established functional studies demonstrating a damaging effect of NM_000535.7:c.2566C>T on PMS2 protein function or splicing were identified.
PP1
No cosegregation data in affected family members were identified for NM_000535.7:c.2566C>T.
PP3
PP3 per PMS2 VCEP v2.0.0 requires either a missense variant with HCI prior probability >0.68, or a predicted splice defect for non-canonical splicing nucleotides with SpliceAI delta >= 0.2.
PP4
No CRC/endometrial MSI-H tumors or loss of MMR protein expression consistent with this variant location were identified for NM_000535.7:c.2566C>T.
Benign
BA1
BA1 per PMS2 VCEP v2.0.0 requires gnomAD v4 grpmax filtering allele frequency >= 0.0028 (0.28%).
BS1
BS1 per PMS2 VCEP v2.0.0 requires gnomAD v4 grpmax filtering allele frequency >= 0.00028 and < 0.0028.
BS2
No reports of NM_000535.7:c.2566C>T observed in trans with a known pathogenic PMS2 variant in a patient with colorectal cancer after age 45 and no evidence of CMMRD were identified.
BS3
No well-established functional studies demonstrating no damaging effect of NM_000535.7:c.2566C>T were identified.
BS4
No dedicated segregation studies demonstrating lack of cosegregation with disease were identified for NM_000535.7:c.2566C>T.
BP5
No tumor pathology data (MSS tumors, BRAF V600E, MLH1 methylation) were identified for NM_000535.7:c.2566C>T.
N/A · 11
PVS1 · PS1 · PS4 · PM1 · PM5 · PM6 · PP2 · PP5 · BP1 · BP2 · BP6
Research & evidence
Population frequency · supports benign
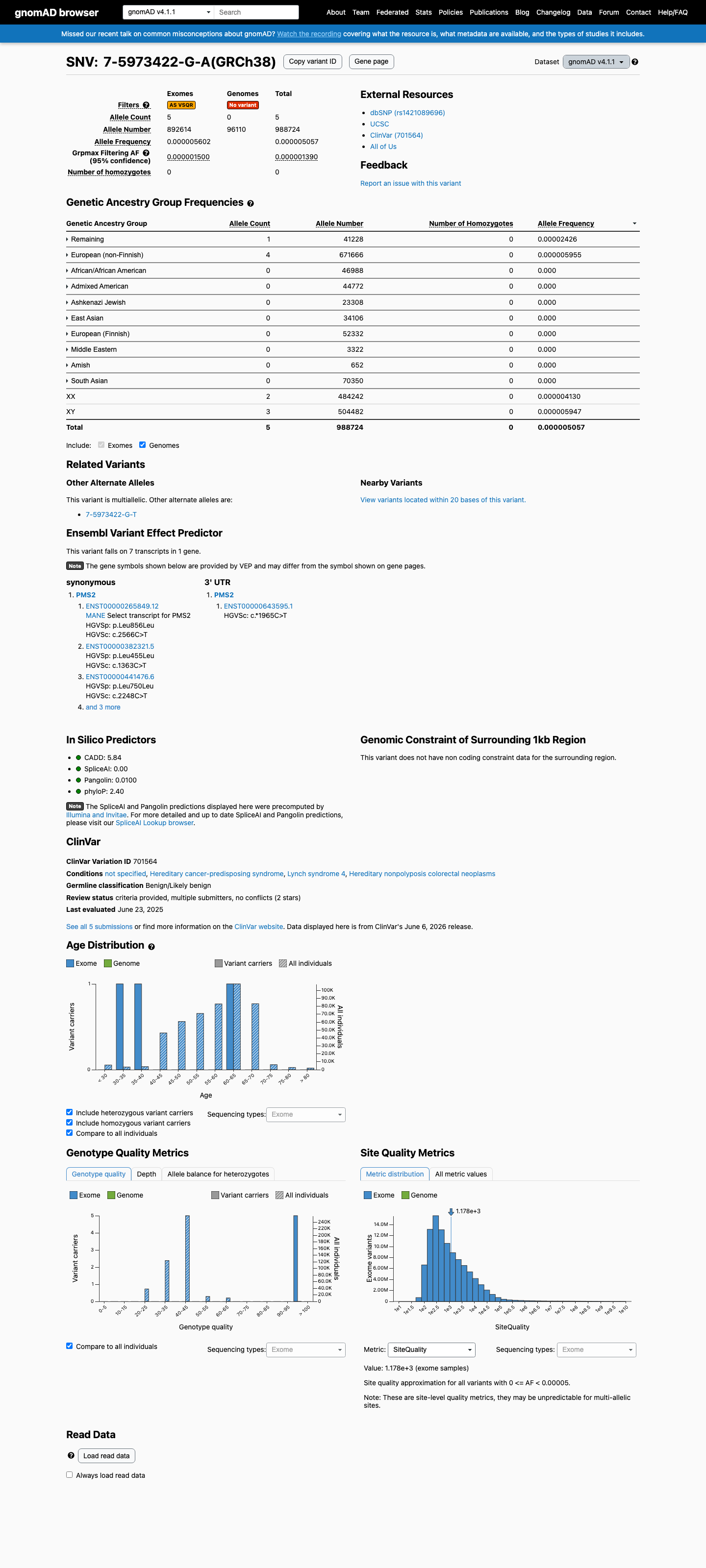
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 5.05702e-06; MAF= 0.00051%, 5/988724 alleles, homozygotes = 0) and has highest observed frequency in the Remaining individuals population (AF= 2.42554e-05; MAF= 0.00243%, 1/41228 alleles, homozygotes = 0); grpmax FAF= 1.39e-06.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00051%
· 5 / 988,724
0 hom · FAF 0.00014%
0 hom · FAF 0.00014%
Remaining individuals 1 / 41,228 |
0.0024% |
European (non-Finnish) 4 / 671,666 |
0.0006% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
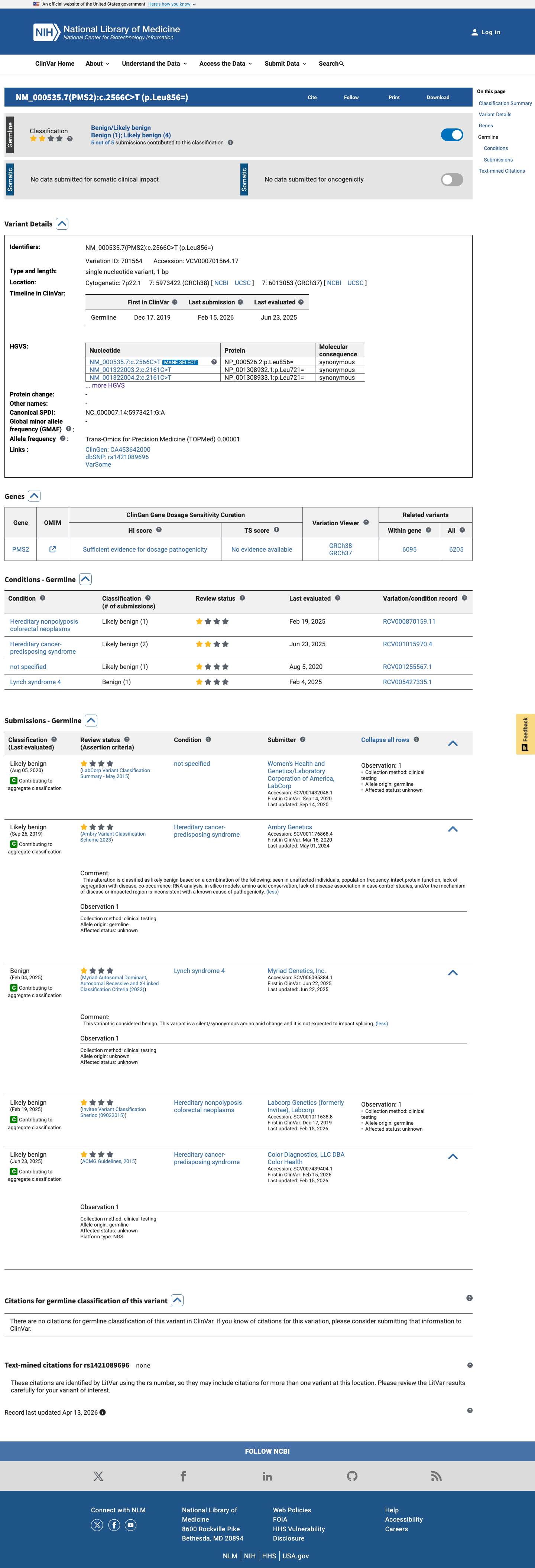
ClinVar
This variant has been reported in ClinVar as Likely benign (4 clinical laboratories) and as Benign (1 clinical laboratory). (ClinVarID = 701564)
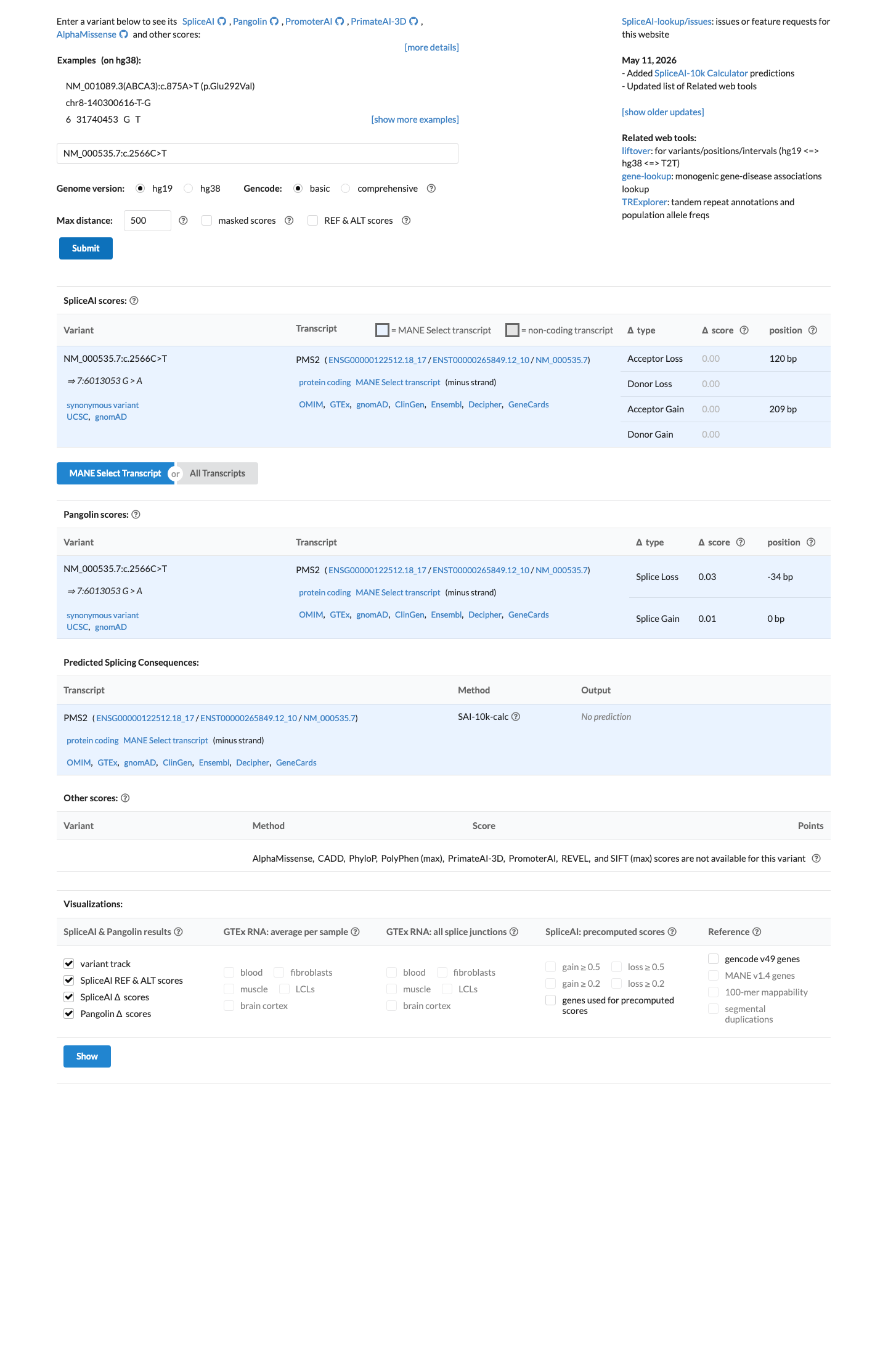
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
23788249 ↗
ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.
CLINVAR
25356965 ↗
ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
27854360 ↗
Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics.
CLINVAR
34012068 ↗
ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR
34043773 ↗
European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR