BA1 (Stand-Alone) is met: the gnomAD v4.1 grpmax filtering allele frequency is 0.408%, exceeding the VCEP BA1 threshold of 0.28%, with one homozygote present in the database, excluding this variant as a cause of Lynch syndrome due to PMS2.1 BP4 (Supporting) is met: the HCI prior probability for pathogenicity is 0.0013, below the BP4_Supporting threshold of <0.11, with multiple in silico predictors (REVEL 0.251, BayesDel -0.187, SpliceAI max delta 0.03) consistent with a benign computational profile.2 This variant has been reported in ClinVar as Benign by 9 clinical laboratories, Likely Benign by 8 laboratories, and Uncertain Significance by 2 laboratories (ClinVar Variation ID: 135946).3 PP3 is not met: the HCI prior of 0.0013 is far below the PP3_Supporting threshold of >0.68.4 PM2_Supporting is not met: the gnomAD v4.1 allele frequency of 0.0239% exceeds the PM2 threshold of <0.002%.5
PMS2
Final classification
Benign
PMS2 c.830C>A · p.Thr277Lys
PMS2
BA1 (Stand-Alone) is met: the gnomAD v4.1 grpmax filtering allele frequency is 0.408%, exceeding the VCEP BA1 threshold of 0.28%, with one homozygote present in the database, excluding this variant as a cause of Lynch syndrome due to PMS2.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PMS2 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule17 (1 Benign.Stand Alone) with applied criteria: BA1 stand-alone benign, BP4 supporting; maps to Benign.
Classification rationale
BA1BP4
Benign
PMS2 c.830C>A
BA1 + BP4
→
Benign
2
hci_priorrevelbayesdelspliceai ↗cspec ↗
4
hci_priorcspec ↗
Gene diagram
· NM_000535.7 · variants mapped to exon structure
PMS2
NM_000535.7
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 13 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
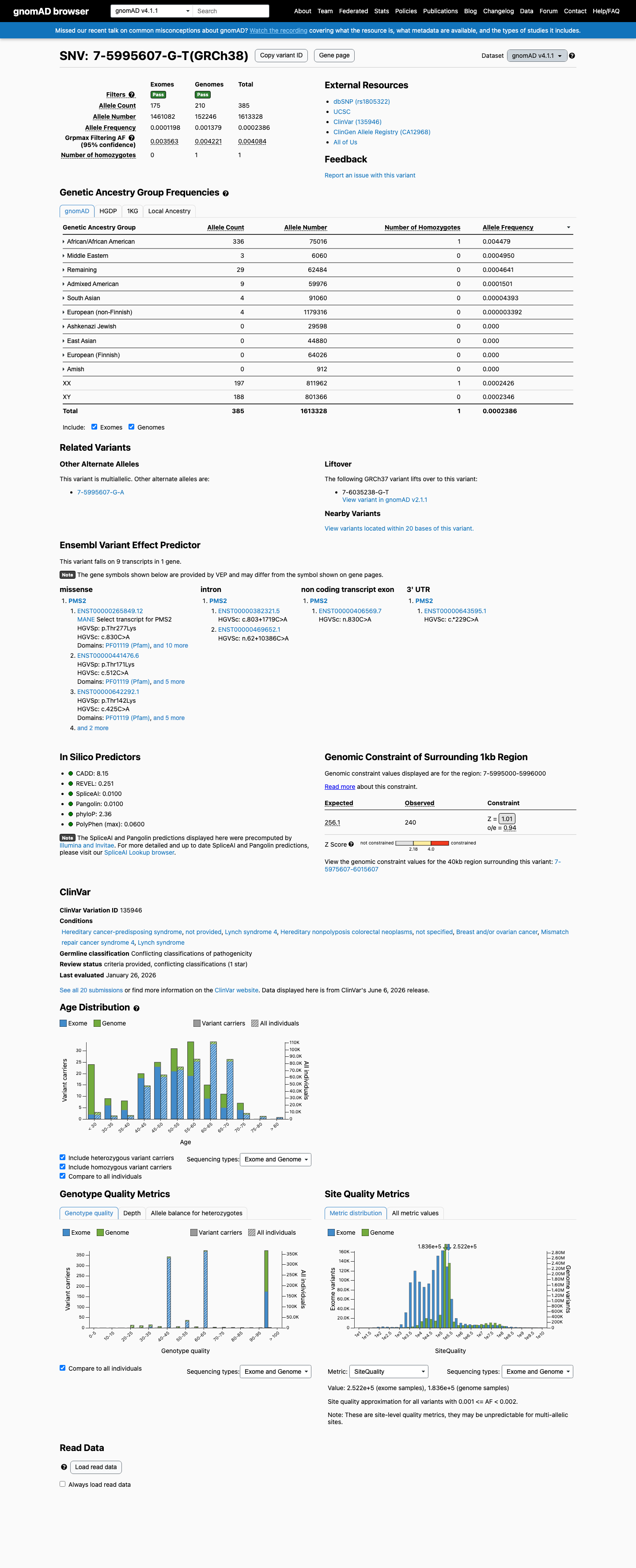
The VCEP BA1 rule is met: gnomAD v4.1 grpmax filtering allele frequency is 0.00408 (0.408%), exceeding the BA1 threshold of ≥0.0028 (0.28%). One homozygote is present in gnomAD v4.1. The variant is enriched in the African/African American population (AF=0.448%) consistent with a population-specific polymorphism rather than a founder pathogenic variant; PMS2 pathogenic variants causing Lynch syndrome are incompatible with a homozygote in a general population database, as homozygosity would cause constitutional mismatch repair deficiency (CMMRD).
gnomAD v4.1: grpmax FAF=0.00408 (0.408%)exceeding BA1 threshold of 0.28%. One homozygote present. African/African American subpopulation AF=0.448%. Absent from gnomAD-Canada v1.0.
✓
BP4
supporting
Benign
The VCEP BP4_Supporting rule is met: the HCI prior probability for c.830C>A (p.Thr277Lys) is 0.0013, which is below the BP4_Supporting threshold of <0.11. Multiple additional in silico predictors support a benign interpretation: REVEL score 0.251, BayesDel score -0.187, and SpliceAI max delta 0.03 (no predicted splicing impact).
HCI prior 0.0013 (<0.11 BP4 threshold). REVEL 0.251. BayesDel -0.187. SpliceAI max delta 0.03. All computational predictors consistent with benign effect.
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change encoding the same amino acid (Thr277Lys) previously classified as Pathogenic by this VCEP.
PS2
No de novo occurrence of NM_000535.7:c.830C>A has been reported in any published study or database.
PS3
No variant-specific functional assay data for NM_000535.7:c.830C>A (p.Thr277Lys) were confirmed from the available evidence.
PM2
The VCEP PM2_Supporting rule requires gnomAD v4 allele frequency <0.00002 (<1 in 50,000 alleles).
PM5
The VCEP PM5 rule requires PP3 to be supporting for the missense change, and a different missense at the same residue (Thr277) classified as Pathogenic or Likely Pathogenic by this VCEP.
PP1
No co-segregation data are available for this variant in families with Lynch syndrome or CMMRD.
PP3
The VCEP PP3 rule uses the HCI prior probability for pathogenicity.
PP4
No tumor MSI/IHC data are available for individuals carrying this variant.
Benign
BS1
The VCEP BS1 threshold is ≥0.00028 (0.028%) and <0.0028 (0.28%).
BS2
No data are available on co-occurrence of this variant in trans with a known pathogenic PMS2 variant in a patient with colorectal cancer after age 45 without CMMRD features.
BS3
No variant-specific functional assay data confirming normal MMR function for NM_000535.7:c.830C>A were identified.
BS4
No family-based lack of co-segregation data are available for this variant.
BP5
No tumor data (MSS CRC/endometrial tumors, retained MMR protein expression, BRAF V600E, or MLH1 methylation) are available for carriers of this variant.
N/A · 11
PVS1 · PS4 · PM1 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6 · BP7
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000238637; MAF= 0.02386%, 385/1613328 alleles, homozygotes = 1) and has highest observed frequency in the African/African American population (AF= 0.00447904; MAF= 0.44790%, 336/75016 alleles, homozygotes = 1); grpmax FAF= 0.00408391.
v2.1
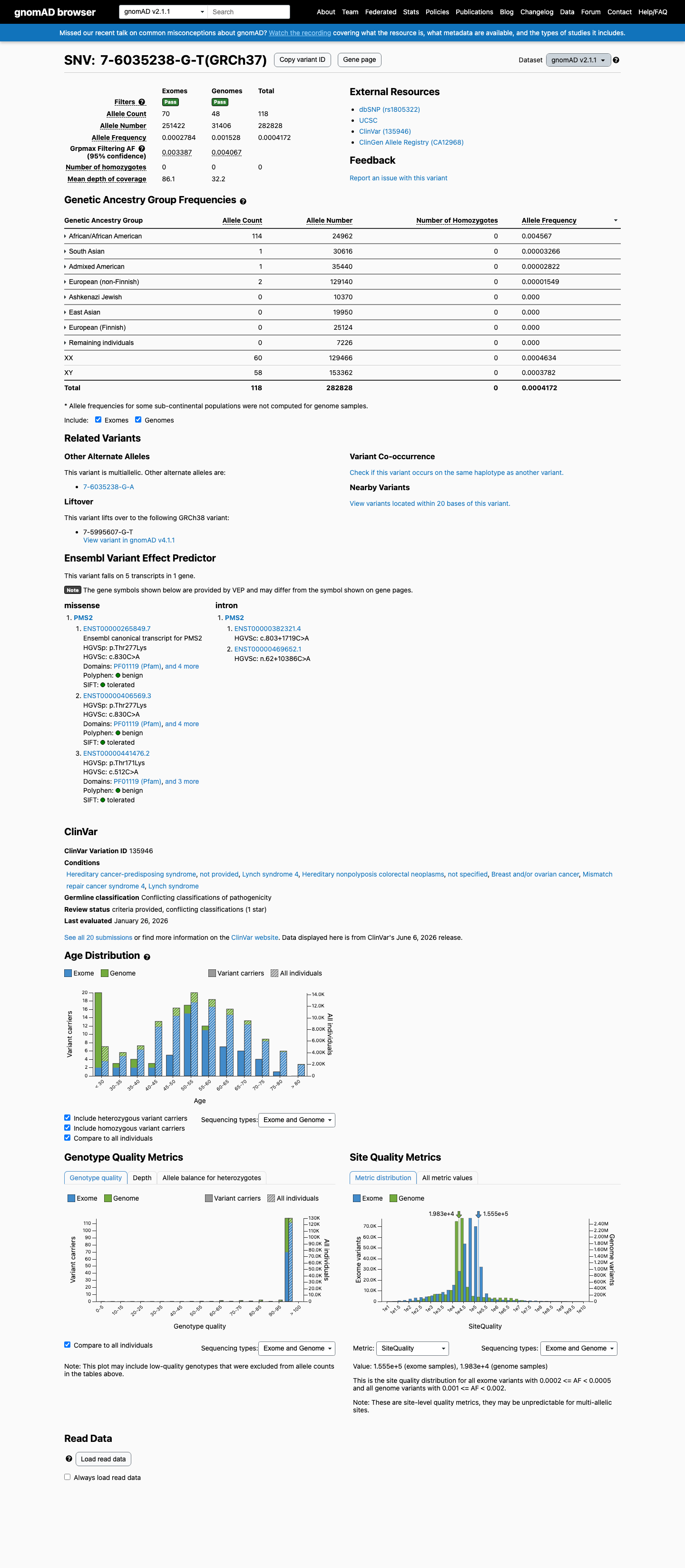
This variant is present in gnomAD v2.1 (AF= 0.000417215; MAF= 0.04172%, 118/282828 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0.00456694; MAF= 0.45669%, 114/24962 alleles, homozygotes = 0); grpmax FAF= 0.00406668.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0001086484137331595, 2/18408 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.024%
· 385 / 1,613,328
1 hom · FAF 0.41%
1 hom · FAF 0.41%
African/African American 336 / 75,016 |
0.45% 1 hom |
Middle Eastern 3 / 6,060 |
0.05% |
Remaining individuals 29 / 62,484 |
0.046% |
Admixed American 9 / 59,976 |
0.015% |
South Asian 4 / 91,060 |
0.0044% |
European (non-Finnish) 4 / 1,179,316 |
0.00034% |
+ 4 not observed (European (Finnish), Amish, East Asian, Ashkenazi Jewish)
gnomAD v2.1
0.042%
· 118 / 282,828
0 hom · FAF 0.41%
0 hom · FAF 0.41%
African/African American 114 / 24,962 |
0.46% |
South Asian 1 / 30,616 |
0.0033% |
Admixed American 1 / 35,440 |
0.0028% |
European (non-Finnish) 2 / 129,140 |
0.0015% |
+ 4 not observed (Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
0.011%
· 2 / 18,408
0 hom · FAF 0.035%
0 hom · FAF 0.035%
African/African American 2 / 1,020 |
0.2% |
+ 8 not observed (Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, European (non-Finnish), Remaining individuals, South Asian)
ClinVar
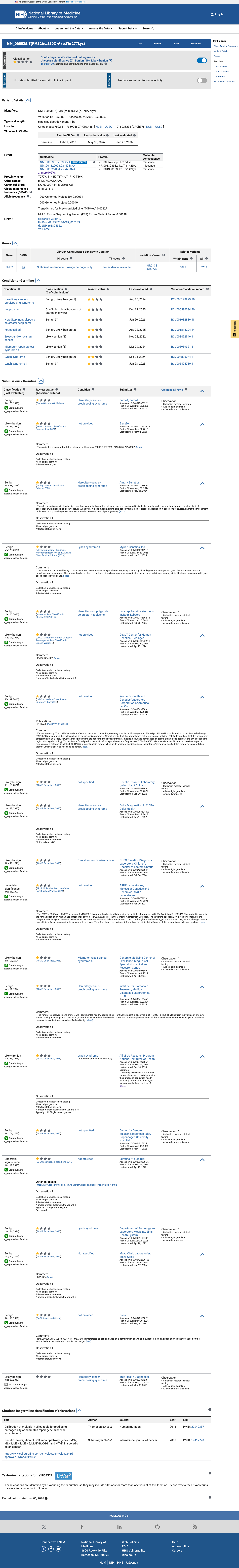
This variant has been reported in ClinVar as Benign (9 clinical laboratories) and as Likely benign (7 clinical laboratories) and as Uncertain significance (2 clinical laboratories) and as Likely Benign (1 clinical laboratory). (ClinVarID = 135946)
In silico
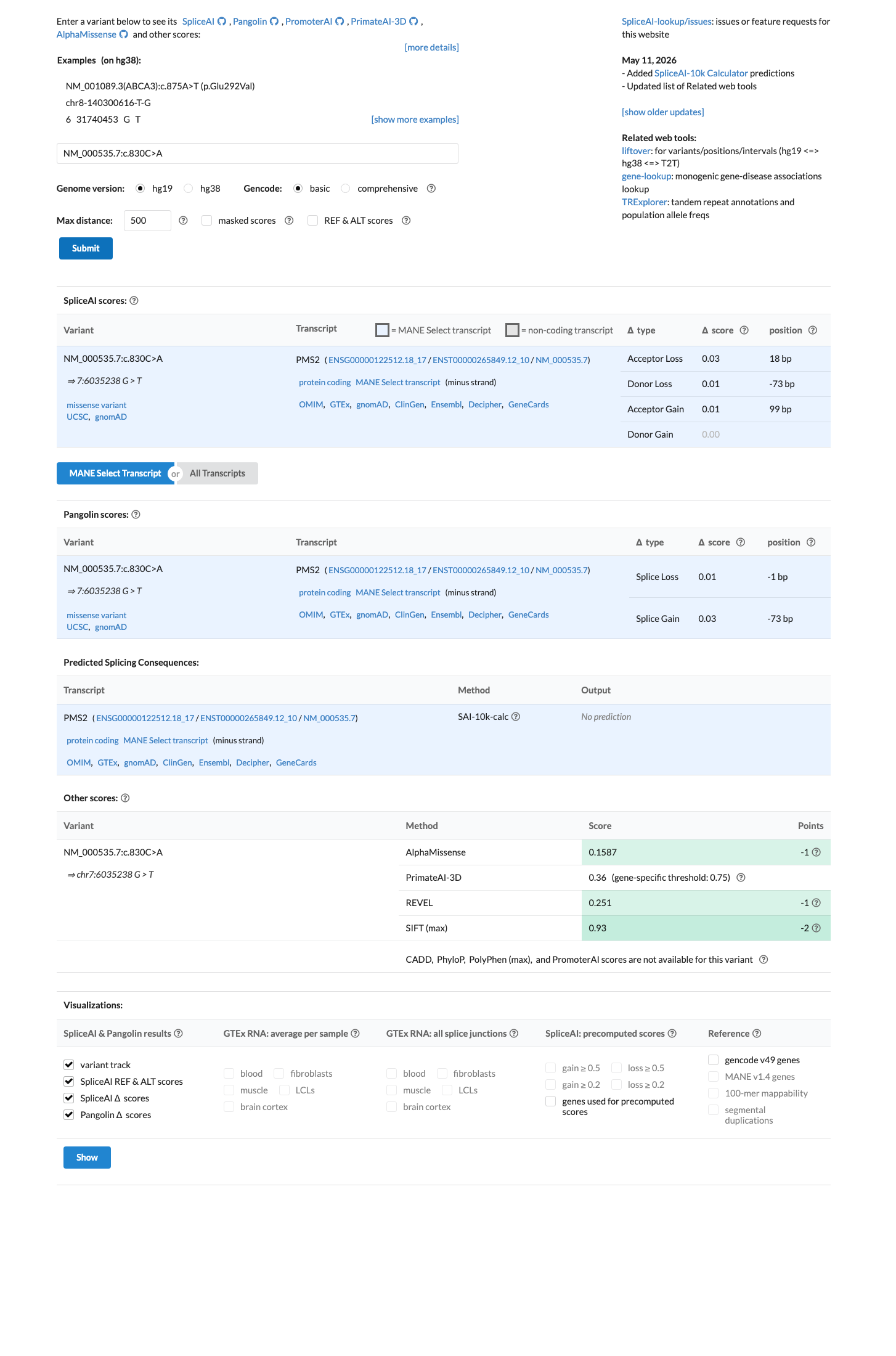
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.03). REVEL score = 0.251. BayesDel score = -0.187245. HCI prior probability for pathogenicity = 0.0013. MAPP score = 3.08. Custom PP2 score = 0.007.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. PMS2, an endonuclease involved in DNA repair, is altered in various cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 8 further PMIDs triaged but not cited — see Sources & References.
Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions.
Searched
c.830C>Ap.Thr277LysT277K
Found
Thompson et al. (2013) calibrated multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. The variant NM_000535.7:c.830C>A (p.Thr277Lys) is included in the supplementary HCI priors table with an MAPP/PP2 prior probability of 0.0013, MAPP score 3.08, and custom PP2 score 0.007.
Variant
✓ Names this variant
Applied to
→BP4 supports · met
Why
Variant-specific HCI prior score of 0.0013 confirms BP4_Supporting is met; same score confirms PP3 is not met.
c.830C>A, p.T277K, custom PP2 score 0.007, MAPP score 3.08, MAPP/PP2 Prior 0.0013
Location Supplementary Table (HCI priors database); variant entry PMS2_01804
Sources & reference links
9Sources
Triaged references · 8 PMIDs not cited in assessment
17417778 ↗
Genetic investigation of DNA-repair pathway genes PMS2, MLH1, MSH2, MSH6, MUTYH, OGG1 and MTH1 in sporadic colon cancer.
CLINVAR
25070057 ↗
Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer.
CLINVAR
25645574 ↗
ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
11598466 ↗
Practice parameters for the identification and testing of patients at risk for dominantly inherited colorectal cancer--supporting documentation.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR