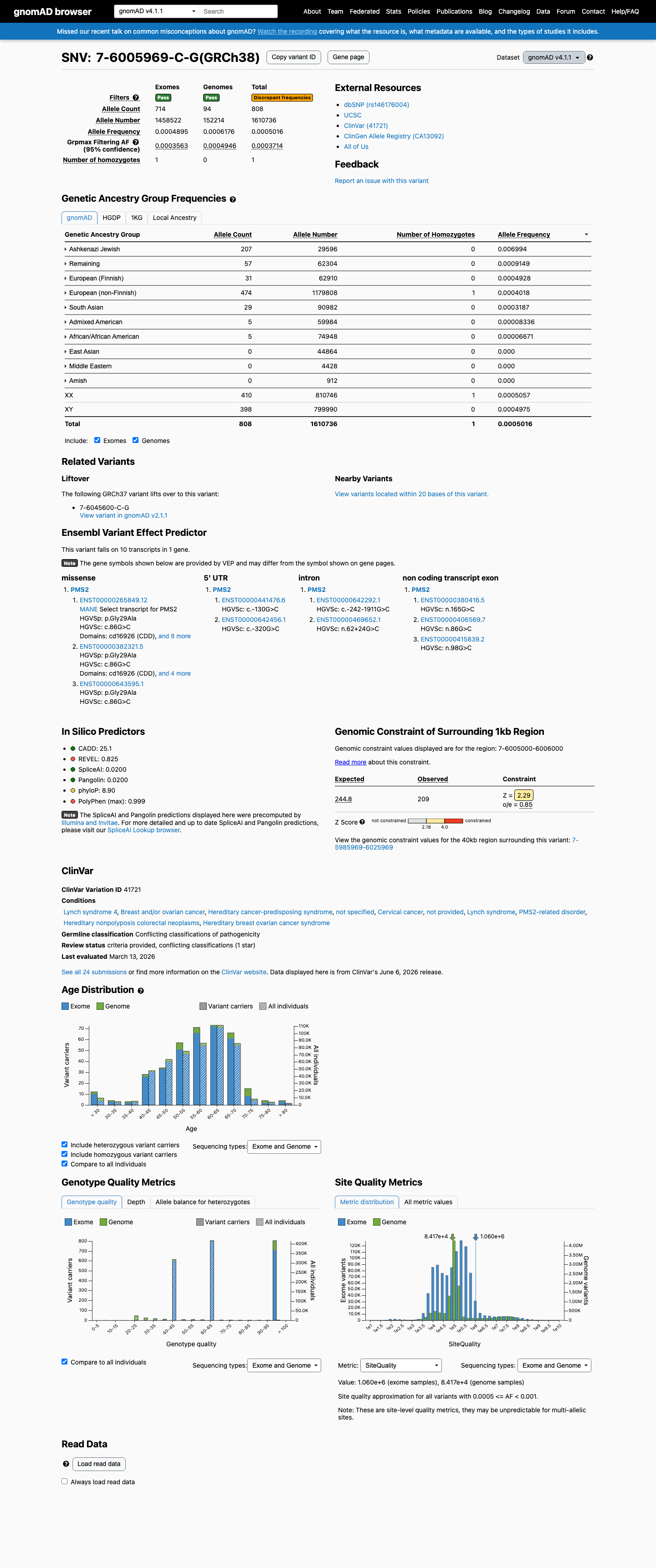
NM_000535.7:c.86G>C (p.Gly29Ala) is a missense variant in exon 2 of PMS2, a gene in which loss-of-function is an established mechanism for Lynch syndrome.1 This variant is present in gnomAD v4.1 at an allele frequency of 0.050% (808/1,610,736 alleles, grpmax FAF=0.037%), which meets the VCEP PMS2 BS1_Strong threshold (grpmax FAF ≥ 0.028% and < 0.28%). One homozygote is observed in both gnomAD v2 and v4, further supporting that this variant is unlikely to be highly penetrant.2 The HCI MAPP/PP2 Prior P score of 0.8883 exceeds the VCEP PP3_Moderate threshold (>0.81), providing in silico evidence for a deleterious effect.3 This variant is reported in ClinVar as Likely benign by 10 clinical laboratories and as Benign by 2 laboratories, though 5 laboratories classify it as Uncertain significance. The overall ClinVar classification is Likely benign with review status of criteria provided, single submitter.4 SpliceAI predicts no splicing impact (max delta score 0.00), and the variant does not lie in a statistically significant mutational hotspot.5 No functional studies, segregation data, tumor phenotype data, or de novo observations are available for this variant. No peer-reviewed publication specifically mentions NM_000535.7:c.86G>C.
PMS2
Final classification
Uncertain Significance - Conflicting Evidence
PMS2 c.86G>C · p.Gly29Ala
PMS2
NM_000535.7:c.86G>C (p.Gly29Ala) is a missense variant in exon 2 of PMS2, a gene in which loss-of-function is an established mechanism for Lynch syndrome.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PMS2 Version 2.0.0 v2.0.0 criteria-combination framework: matched Rule22 (Benign.Strong >=1 + Pathogenic.Moderate >=1) with applied criteria: PP3 moderate, BS1 strong; maps to Uncertain Significance - Conflicting Evidence.
Classification rationale
PP3
BS1
Uncertain Significance - Conflicting Evidence
PMS2 c.86G>C
PP3 + BS1
→
Uncertain Significance - Conflicting Evidence
Gene diagram
· NM_000535.7 · variants mapped to exon structure
PMS2
NM_000535.7
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 13 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PP3
moderate
Pathogenic
The HCI MAPP/PP2 Prior P score for p.Gly29Ala (PMS2_00179) is 0.8883, which exceeds the VCEP PP3_Moderate threshold of >0.81. This in silico prior probability supports a pathogenic computational prediction.
HCI prior probability = 0.8883 (MAPP score 22.05custom PP2 score 0.998). Exceeds VCEP PP3_Moderate threshold (>0.81).
✓
BS1
strong
Benign
Per VCEP PMS2 rule, BS1_Strong applies when gnomAD v4 grpmax filtering allele frequency is ≥ 0.00028 (0.028%) and < 0.0028 (0.28%), and the variant is excluded as a founder pathogenic variant. The grpmax FAF for this variant is 0.00037135 (0.037%), which falls within this range. The variant is observed across multiple populations with highest frequency in Ashkenazi Jewish (0.699%), but there is no definitive evidence of founder pathogenic status. One homozygote is observed in gnomAD v4, further supporting that this variant is unlikely to be highly penetrant.
gnomAD v4 grpmax FAF = 0.00037135 (between 0.00028 and 0.0028 VCEP BS1 range). 808 alleles observedincluding 1 homozygote. Highest subpopulation frequency in Ashkenazi Jewish (AF=0.00699).
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change producing the same amino acid change (p.Gly29Ala) that has been classified as Pathogenic or Likely Pathogenic by this VCEP.
PS2
No de novo observations have been reported for NM_000535.7:c.86G>C in any available data source.
PS3
No variant-specific functional assay data exist for NM_000535.7:c.86G>C (p.Gly29Ala).
PM2
Per VCEP PMS2 rule, PM2_Supporting requires absence or extremely rare allele frequency <0.00002 (<1 in 50,000 alleles) in gnomAD v4.
PM5
PM5 requires a different missense change at amino acid residue Gly29 classified as Pathogenic or Likely Pathogenic by this VCEP.
PP1
No co-segregation data are available for NM_000535.7:c.86G>C.
PP4
No tumor MSI/IHC data are available for NM_000535.7:c.86G>C carriers.
Benign
BA1
Per VCEP PMS2 rule, BA1 requires gnomAD v4 grpmax filtering allele frequency ≥ 0.0028 (0.28%).
BS2
No evidence of co-occurrence in trans with a known pathogenic PMS2 variant in a patient with colorectal cancer after age 45 without CMMRD features.
BS3
No functional assay data demonstrating normal MMR function for NM_000535.7:c.86G>C exist.
BS4
No co-segregation data are available to demonstrate lack of segregation with disease.
BP4
Per VCEP PMS2 rule, BP4_Supporting requires an HCI MAPP/PP2 Prior P score <0.11.
BP5
No tumor data (MSS status, MMR protein expression) are available for carriers of this variant.
N/A · 11
PVS1 · PS4 · PM1 · PM6 · PP2 · PP5 · BP1 · BP2 · BP3 · BP6 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000501634; MAF= 0.05016%, 808/1610736 alleles, homozygotes = 1) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00699419; MAF= 0.69942%, 207/29596 alleles, homozygotes = 0); grpmax FAF= 0.00037135.
v2.1
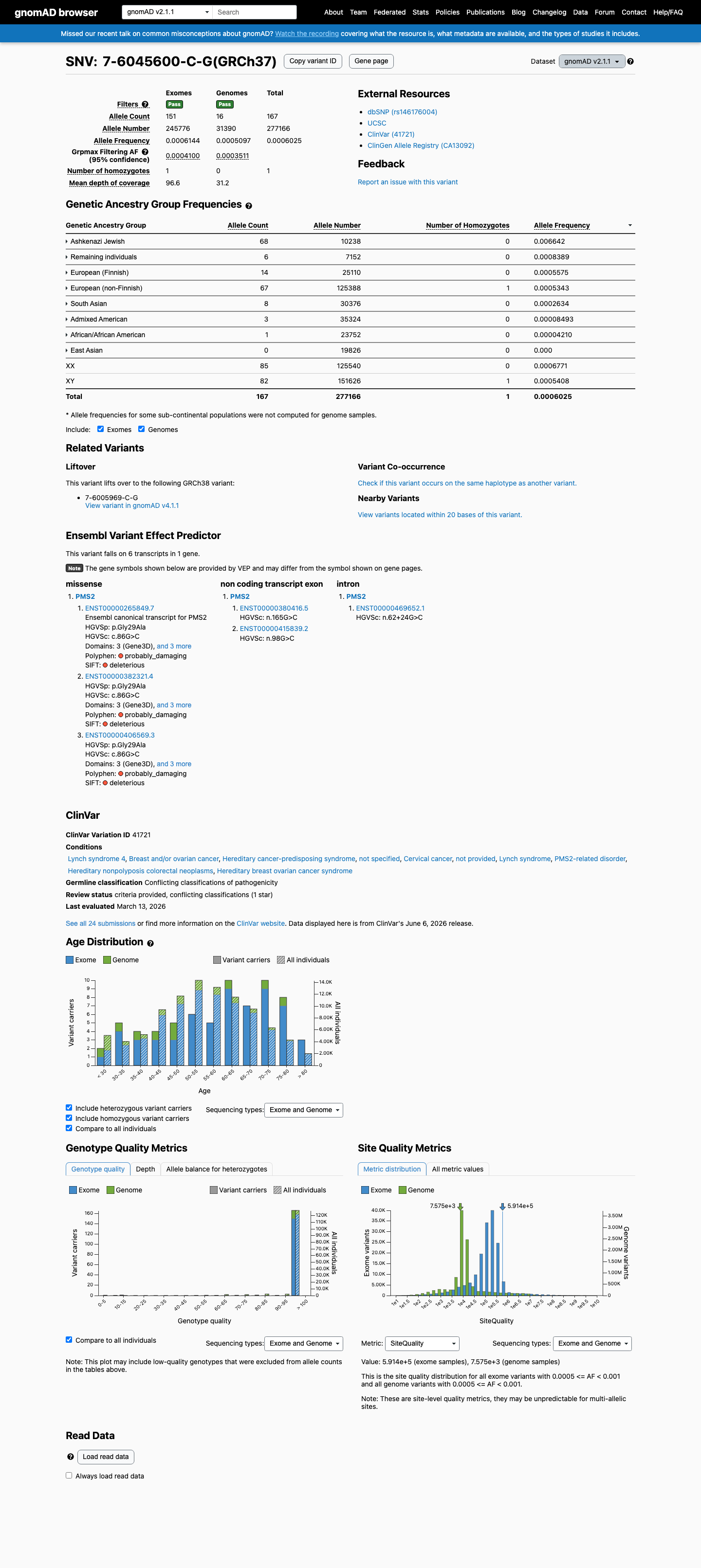
This variant is present in gnomAD v2.1 (AF= 0.000602527; MAF= 0.06025%, 167/277166 alleles, homozygotes = 1) and has highest observed frequency in the Ashkenazi Jewish population (AF= 0.00664192; MAF= 0.66419%, 68/10238 alleles, homozygotes = 0); grpmax FAF= 0.00040995.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.000488599348534202, 9/18420 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.05%
· 808 / 1,610,736
1 hom · FAF 0.037%
1 hom · FAF 0.037%
Ashkenazi Jewish 207 / 29,596 |
0.7% |
Remaining individuals 57 / 62,304 |
0.091% |
European (Finnish) 31 / 62,910 |
0.049% |
European (non-Finnish) 474 / 1,179,808 |
0.04% 1 hom |
South Asian 29 / 90,982 |
0.032% |
Admixed American 5 / 59,984 |
0.0083% |
African/African American 5 / 74,948 |
0.0067% |
+ 3 not observed (Amish, East Asian, Middle Eastern)
gnomAD v2.1
0.06%
· 167 / 277,166
1 hom · FAF 0.041%
1 hom · FAF 0.041%
Ashkenazi Jewish 68 / 10,238 |
0.66% |
Remaining individuals 6 / 7,152 |
0.084% |
European (Finnish) 14 / 25,110 |
0.056% |
European (non-Finnish) 67 / 125,388 |
0.053% 1 hom |
South Asian 8 / 30,376 |
0.026% |
Admixed American 3 / 35,324 |
0.0085% |
African/African American 1 / 23,752 |
0.0042% |
+ 1 not observed (East Asian)
gnomAD Canada 🇨🇦
0.049%
· 9 / 18,420
0 hom · FAF 0.0069%
0 hom · FAF 0.0069%
Ashkenazi Jewish 5 / 832 |
0.6% |
Remaining individuals 1 / 1,138 |
0.088% |
European (non-Finnish) 3 / 11,740 |
0.026% |
+ 6 not observed (African/African American, Latino/Admixed American, East Asian, European (Finnish), Middle Eastern, South Asian)
ClinVar
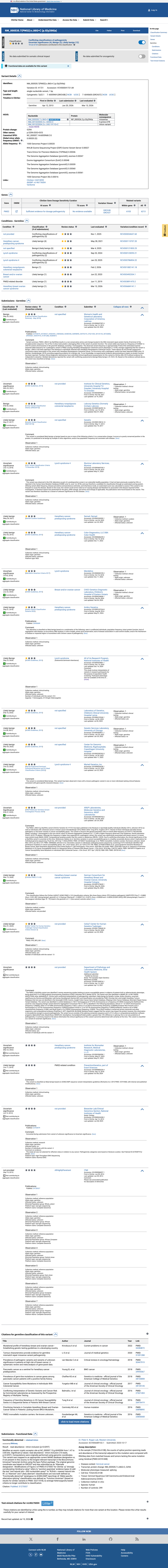
This variant has been reported in ClinVar as Likely benign (10 clinical laboratories) and as Uncertain significance (5 clinical laboratories) and as Benign (2 clinical laboratories) and as Likely Benign (1 clinical laboratory) and as Uncertain Significance (1 clinical laboratory). (ClinVarID = 41721)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.825. BayesDel score = 0.346178. HCI prior probability for pathogenicity = 0.8883. MAPP score = 22.05. Custom PP2 score = 0.998.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. PMS2, an endonuclease involved in DNA repair, is altered in various cancers.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 9 PMIDs not cited in assessment
25070057 ↗
Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
26898890 ↗
Prioritizing Variants in Complete Hereditary Breast and Ovarian Cancer Genes in Patients Lacking Known BRCA Mutations.
CLINVAR
31391288 ↗
Tumour characteristics provide evidence for germline mismatch repair missense variant pathogenicity.
CLINVAR
34284872 ↗
Mutational profile of hereditary breast and ovarian cancer - Establishing genetic testing guidelines in a developing country.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR