Classification rationale
BS1BP4
Likely Benign
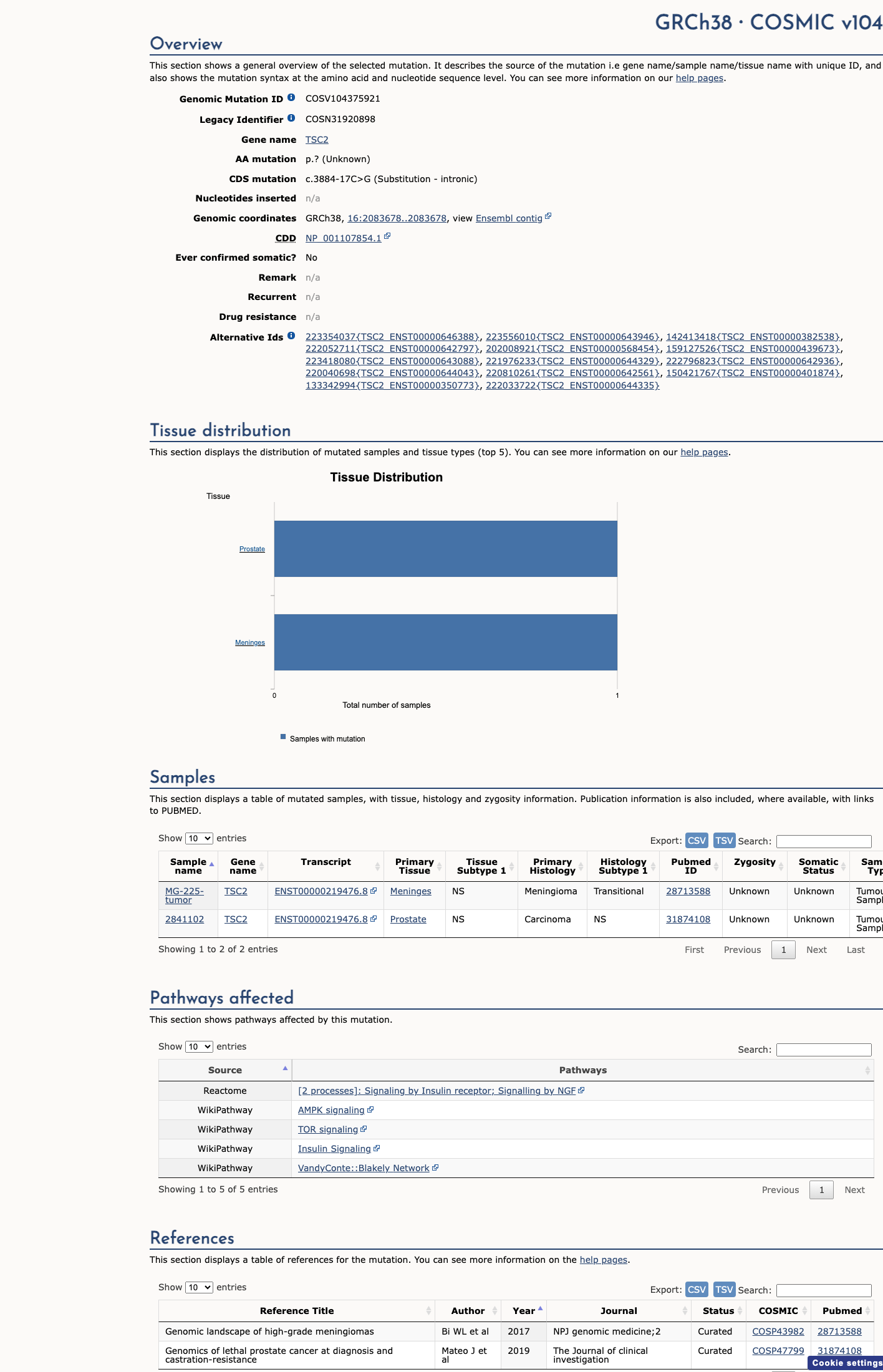
TSC2 c.3884-17C>G
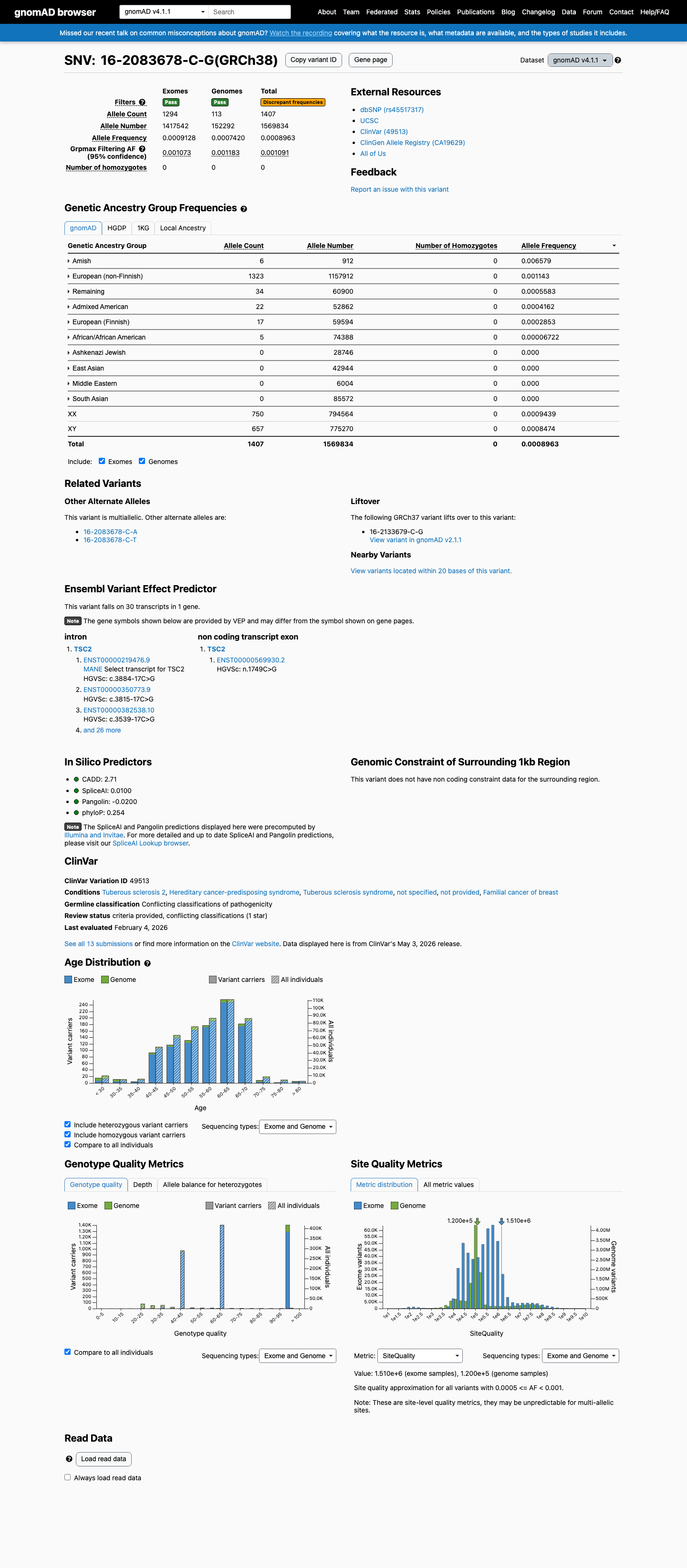
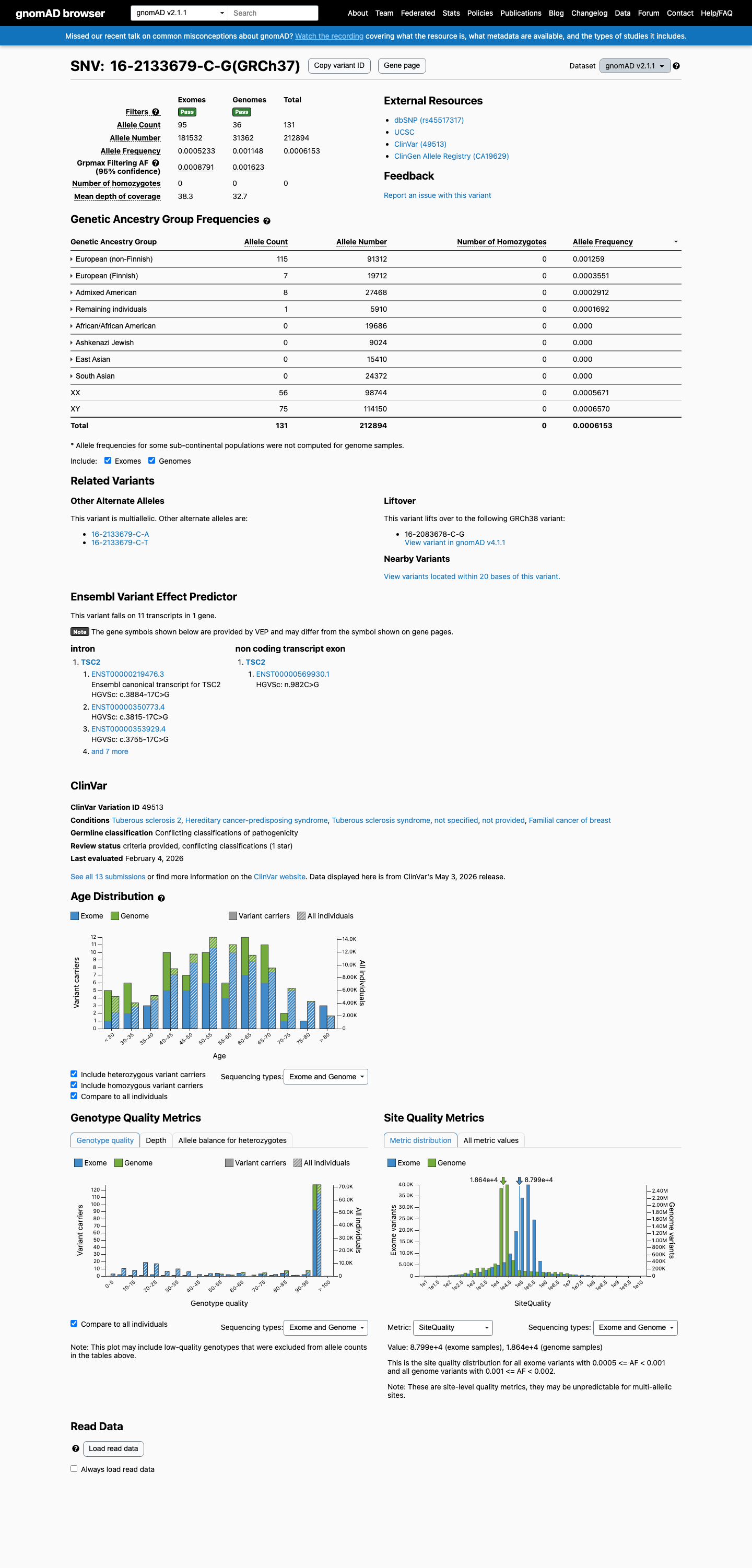
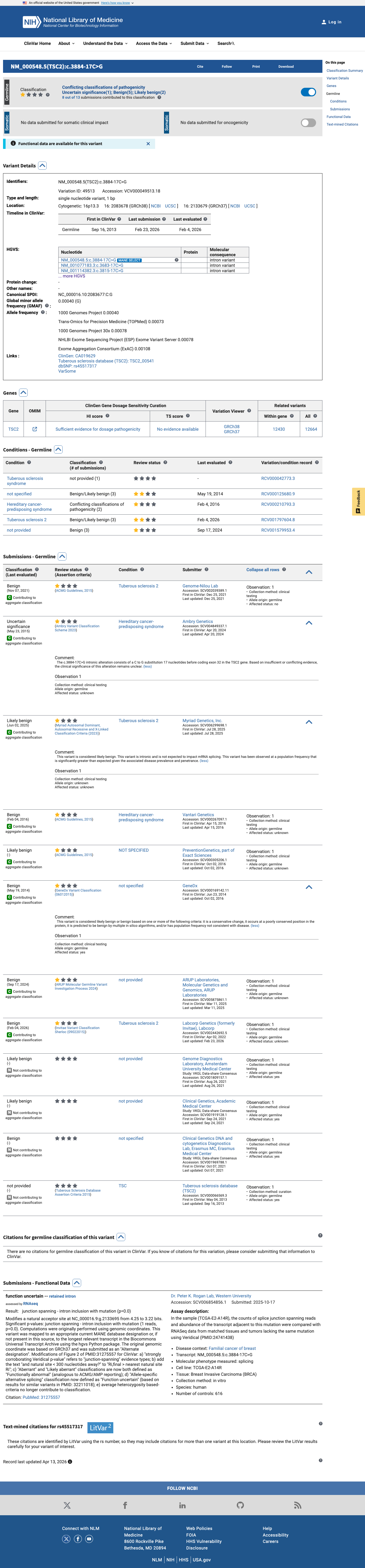
The TSC2 NM_000548.5:c.3884-17C>G (NP_000539.2:p.?) variant has been reported in ClinVar predominantly as benign or likely benign, without an expert panel review.1 This variant is present in population databases, including gnomAD v2.1, gnomAD v4.1, and gnomAD-Canada, with the highest observed frequency of 0.65789% (6/912) in the Amish population in gnomAD v4.1, which is above the default BS1 threshold of 0.3% and below the default BA1 threshold of 1%.2 In silico splicing analysis does not support a deleterious effect, as SpliceAI predicts no significant splice impact with a maximum delta score of 0.00.3
BS1 + BP4
→
Likely Benign