NM_000548.5:c.5186G>A (p.Arg1729His) in TSC2 is a missense variant in exon 41. PVS1 is not applicable as this is not a null variant.1 This variant is present at very low frequency in gnomAD v2.1 (AF=0.00607%, 17/280,130 alleles) and gnomAD v4.1 (AF=0.00355%, 57/1,604,608 alleles), with no homozygotes observed, meeting PM2 at supporting strength.2 This variant has been classified as Likely benign by three clinical laboratories (GeneDx, Ambry Genetics, Labcorp Genetics) and as Benign by two clinical laboratories in ClinVar (variation ID 207786), meeting BP6 at supporting benign strength.3 In silico predictions are conflicting: REVEL score is 0.811 (damaging) while BayesDel score is 0.179 (benign) and SpliceAI predicts no splice impact (max delta 0.00). Neither PP3 nor BP4 can be applied due to mixed computational evidence.4 No variant-specific functional studies, de novo observations, cosegregation data, or case-control evidence were identified for NM_000548.5:c.5186G>A in the reviewed literature. OncoKB reports Unknown Oncogenic Effect.5 No pathogenic classification was reported by any ClinVar submitter. PS5 and PP5 are not met.6 Allele frequency does not meet BA1 (>1%) or BS1 (>0.3%) thresholds. No homozygotes have been observed, so BS2 is not met.7 With PM2 (supporting pathogenic) and BP6 (supporting benign) as the only scorable criteria, these opposing supporting-level criteria effectively neutralize each other. The variant remains a Variant of Uncertain Significance under generic ACMG/AMP 2015 rules.8
TSC2
Final classification
VUS
TSC2 c.5186G>A · p.Arg1729His
TSC2
NM_000548.5:c.5186G>A (p.Arg1729His) in TSC2 is a missense variant in exon 41. PVS1 is not applicable as this is not a null variant.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP6 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP6
VUS
TSC2 c.5186G>A
PM2 + BP6
→
VUS
Gene diagram
· NM_000548.5 · variants mapped to exon structure
TSC2
NM_000548.5
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
NM_000548.5:c.5186G>A is present at very low frequency in population databases: gnomAD v2.1 allele frequency 0.00607% (17/280,130 alleles, 0 homozygotes) and gnomAD v4.1 allele frequency 0.00355% (57/1,604,608 alleles, 0 homozygotes). Both frequencies are below the 0.1% PM2 threshold for rare disease variants. Absent from gnomAD-Canada v1.0.
gnomAD v2.1: AF=0.00607% (17/280130 alleles). gnomAD v4.1: AF=0.00355% (57/1604
✓
BP6
supporting
Benign
NM_000548.5:c.5186G>A has been classified as Likely benign by GeneDx, Ambry Genetics, and Labcorp Genetics (formerly Invitae), and as Benign by Genome-Nilou Lab and an additional clinical laboratory, for a total of 5 clinical diagnostic laboratories reporting benign or likely benign without shared internal data. This meets BP6 at supporting benign strength.
ClinVar variation 207786: Likely benign by 3 clinical laboratories (GeneDxAmbry GeneticsLabcorp Genetics)
Assessed · not applied
Pathogenic
PS1
No same-amino-acid change comparator variant with established pathogenicity was identified for p.Arg1729His.
PS2
No de novo observation data were identified for NM_000548.5:c.5186G>A in any reviewed source.
PS3
No variant-specific functional evidence was identified for p.Arg1729His.
PS4
No case-control or cohort data reporting NM_000548.5:c.5186G>A prevalence in affected individuals were identified.
PM1
Residue p.Arg1729 does not lie within a statistically significant cancer hotspot.
PM6
No de novo observation data were identified for NM_000548.5:c.5186G>A.
PP1
No cosegregation data were identified for NM_000548.5:c.5186G>A in any reviewed source.
PP2
Insufficient data to assess TSC2 missense constraint.
PP3
In silico predictions are conflicting.
PP4
No patient phenotype or family history data were available for NM_000548.5:c.5186G>A to assess specificity for tuberous sclerosis complex.
PP5
No reputable source has reported NM_000548.5:c.5186G>A as pathogenic.
Benign
BA1
NM_000548.5:c.5186G>A allele frequency in gnomAD v2.1 (0.00607%) and v4.1 (0.00355%) is far below the 1% BA1 threshold.
BS1
NM_000548.5:c.5186G>A allele frequency in gnomAD v2.1 (0.00607%) and v4.1 (0.00355%) is far below the 0.3% BS1 threshold.
BS2
No homozygotes for NM_000548.5:c.5186G>A are observed in gnomAD v2.1 or v4.1.
BS3
No well-established functional studies demonstrating no deleterious effect were identified for NM_000548.5:c.5186G>A.
BS4
No cosegregation data were available to assess lack of segregation with disease for NM_000548.5:c.5186G>A.
BP1
TSC2 is associated with tuberous sclerosis complex through both loss-of-function and missense pathogenic variants.
BP2
No data on whether NM_000548.5:c.5186G>A has been observed in trans with a known pathogenic TSC2 variant.
BP4
In silico predictions are conflicting.
BP5
No data were available on whether NM_000548.5:c.5186G>A was observed in a case with an alternative molecular basis for disease.
N/A · 3
PVS1 · PM5 · BP7
Research & evidence
Population frequency
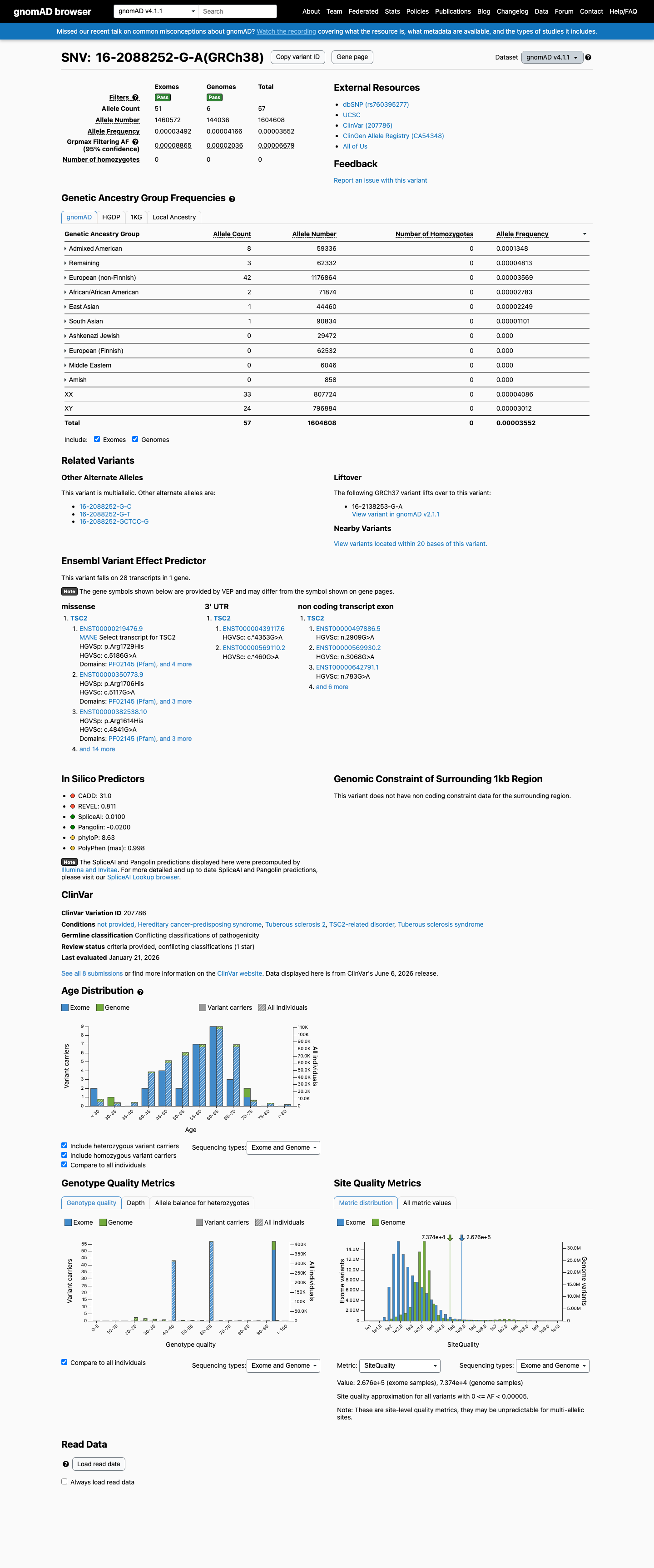
gnomAD v4.1
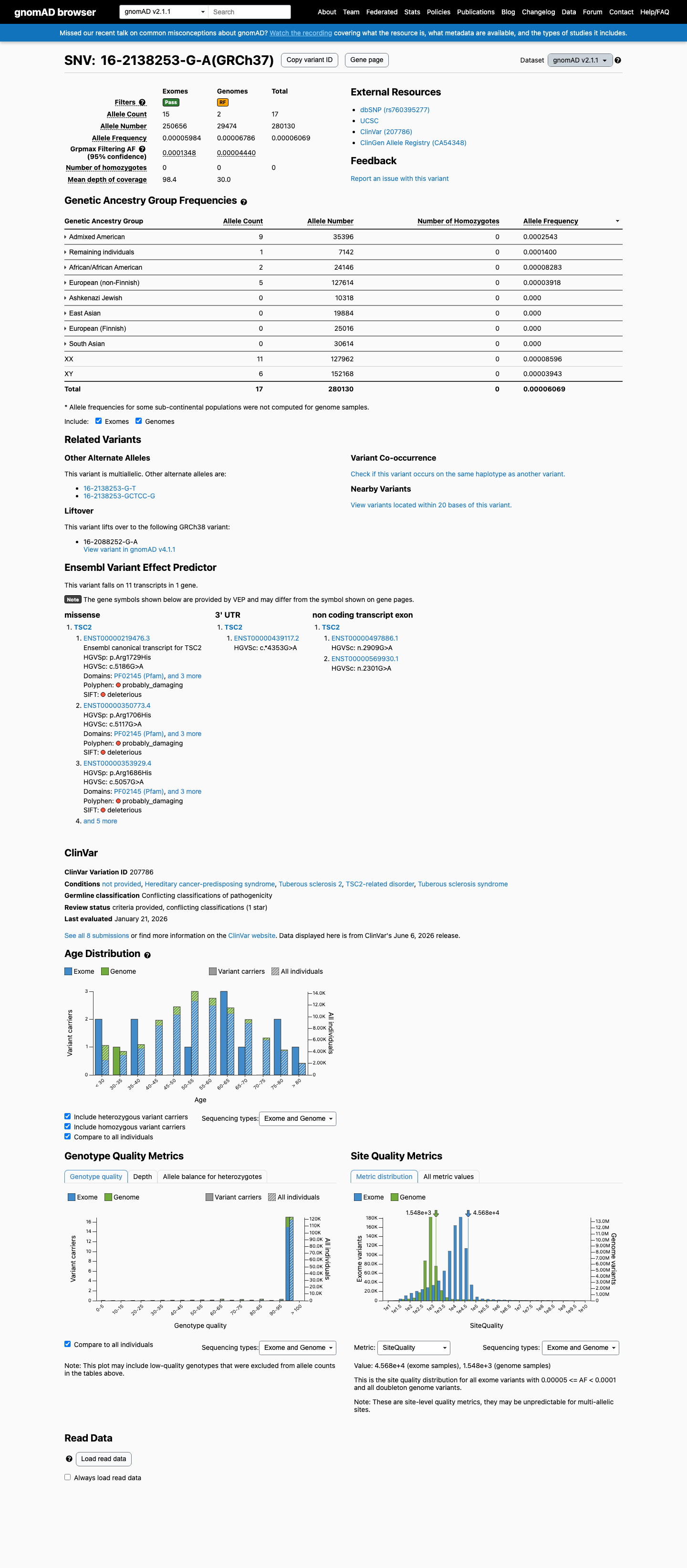
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 3.55227e-05; MAF= 0.00355%, 57/1604608 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000134825; MAF= 0.01348%, 8/59336 alleles, homozygotes = 0); grpmax FAF= 6.679e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 6.06861e-05; MAF= 0.00607%, 17/280130 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 0.000254266; MAF= 0.02543%, 9/35396 alleles, homozygotes = 0); grpmax FAF= 0.00013485.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0036%
· 57 / 1,604,608
0 hom · FAF 0.0067%
0 hom · FAF 0.0067%
Admixed American 8 / 59,336 |
0.013% |
Remaining individuals 3 / 62,332 |
0.0048% |
European (non-Finnish) 42 / 1,176,864 |
0.0036% |
African/African American 2 / 71,874 |
0.0028% |
East Asian 1 / 44,460 |
0.0022% |
South Asian 1 / 90,834 |
0.0011% |
+ 4 not observed (European (Finnish), Amish, Middle Eastern, Ashkenazi Jewish)
gnomAD v2.1
0.0061%
· 17 / 280,130
0 hom · FAF 0.013%
0 hom · FAF 0.013%
Admixed American 9 / 35,396 |
0.025% |
Remaining individuals 1 / 7,142 |
0.014% |
African/African American 2 / 24,146 |
0.0083% |
European (non-Finnish) 5 / 127,614 |
0.0039% |
+ 4 not observed (Ashkenazi Jewish, East Asian, European (Finnish), South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
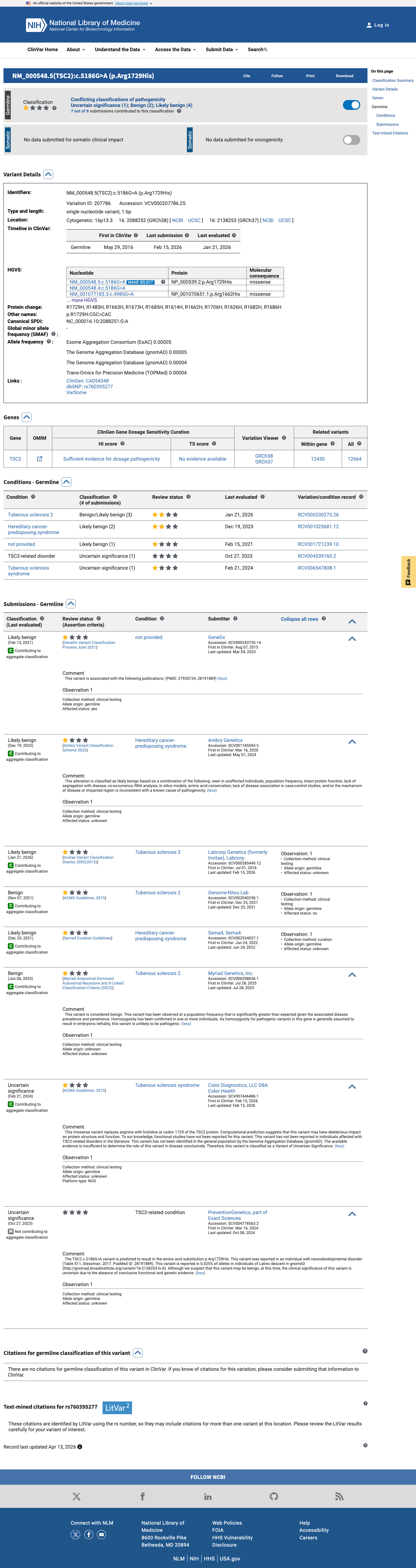
ClinVar
This variant has been reported in ClinVar as Likely benign (3 clinical laboratories) and as Benign (2 clinical laboratories) and as Uncertain significance (2 clinical laboratories). (ClinVarID = 207786)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.811. BayesDel score = 0.178539.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. TSC2, a GTPase-activating protein, is altered by mutation in various cancers, including endometrial and colorectal cancer.
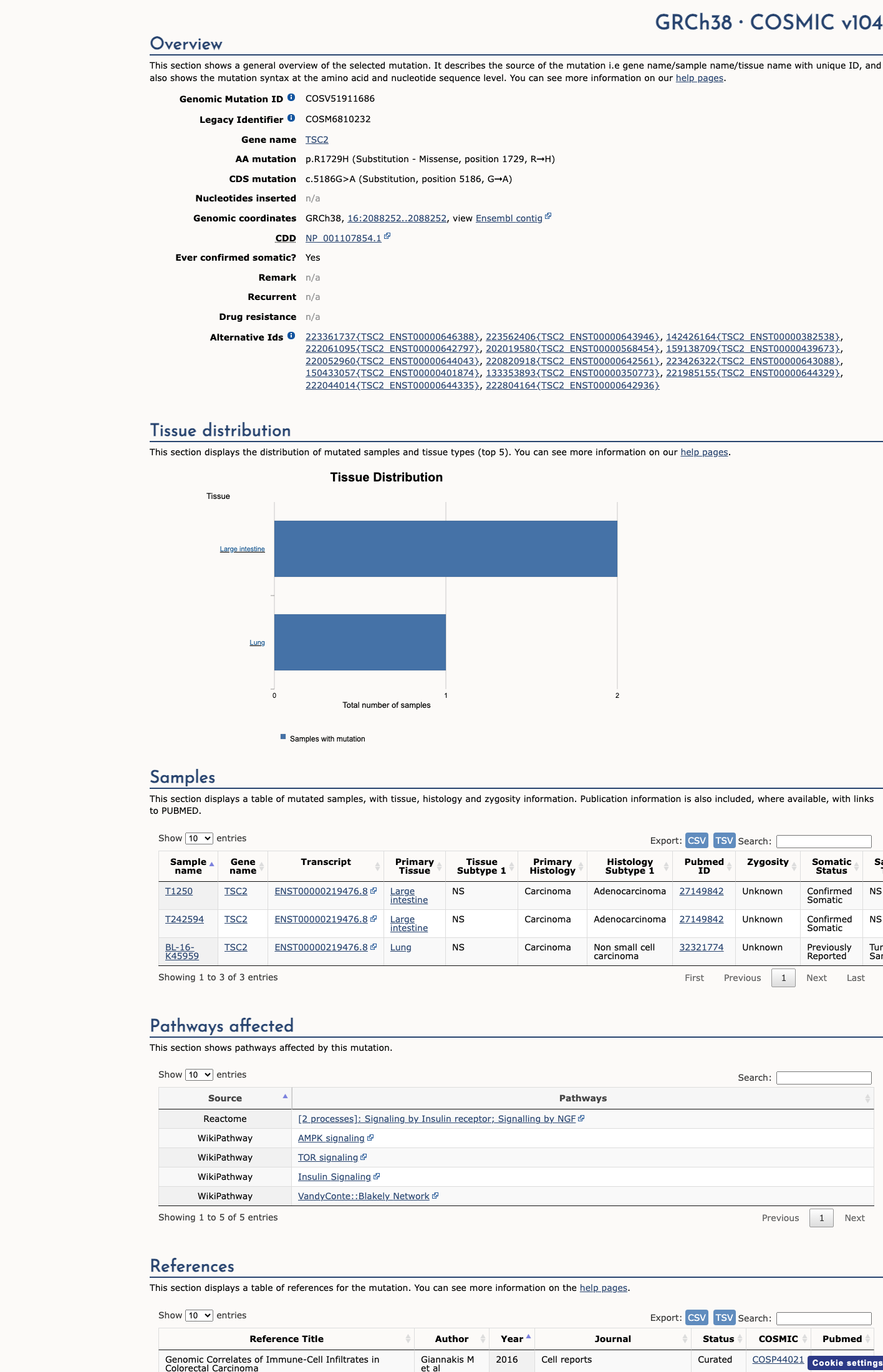
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV51911686, n = 3 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
23519317 ↗
Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions.
CLINVAR
23788249 ↗
ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.
CLINVAR
25356965 ↗
ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing.
CLINVAR
25394175 ↗
A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment.
CLINVAR
27854360 ↗
Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics.
CLINVAR
34012068 ↗
ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR