NM_000551.3:c.104C>T (p.Ala35Val) is a missense variant in exon 1 of VHL, located in the N-terminal region prior to the p19 Met54 initiation site and outside all critical functional domains (Beta: AA63-154, Alpha: AA156-192, Second Beta: AA193-204).1 This variant is present in gnomAD v4.1 at 16 of 1,544,318 alleles (allele frequency 1.04e-5) with a GroupMax Filtering Allele Frequency of 0.01103% (0.0001103) in the South Asian population, exceeding the VHL VCEP BS1 threshold of ≥0.00156% (BS1_Strong).2 SpliceAI predicts no splicing impact (max delta score = 0.00), meeting the VHL VCEP BP4 criterion for lack of splice effect at Supporting strength.3 REVEL score is 0.307, below the VHL VCEP PP3 threshold of ≥0.664. BayesDel score is -0.264 (benign direction). In silico predictors do not support pathogenicity.4 No variant-specific functional data, segregation data, de novo observations, or proband phenotype data are available. The variant has not been reported in COSMIC and does not lie in a statistically significant mutational hotspot. ClinVar classifies this variant as Uncertain significance (variation ID 526678) based on 3 clinical laboratory submissions with no expert panel review.5 Applying the VHL VCEP v1.1.0 classification rules: BS1_Strong (1 strong benign) + BP4_Supporting (1 supporting benign) meets Rule 18 criteria for Likely Benign.6
VHL
Final classification
Likely Benign
VHL c.104C>T · p.Ala35Val
VHL
NM_000551.3:c.104C>T (p.Ala35Val) is a missense variant in exon 1 of VHL, located in the N-terminal region prior to the p19 Met54 initiation site and outside all critical functional domains (Beta: AA63-154, Alpha: AA156-192, Second Beta: AA193-204).
ClinGen VHL Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for VHL Version 1.1.0 v1.1.0 criteria-combination framework: matched Rule18 (1 Benign.Strong + 1 Benign.Supporting) with applied criteria: BS1 strong, BP4 supporting; maps to Likely Benign.
Classification rationale
BS1BP4
Likely Benign
VHL c.104C>T
BS1 + BP4
→
Likely Benign
Gene diagram
· NM_000551.3 · variants mapped to exon structure
VHL
NM_000551.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 17 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
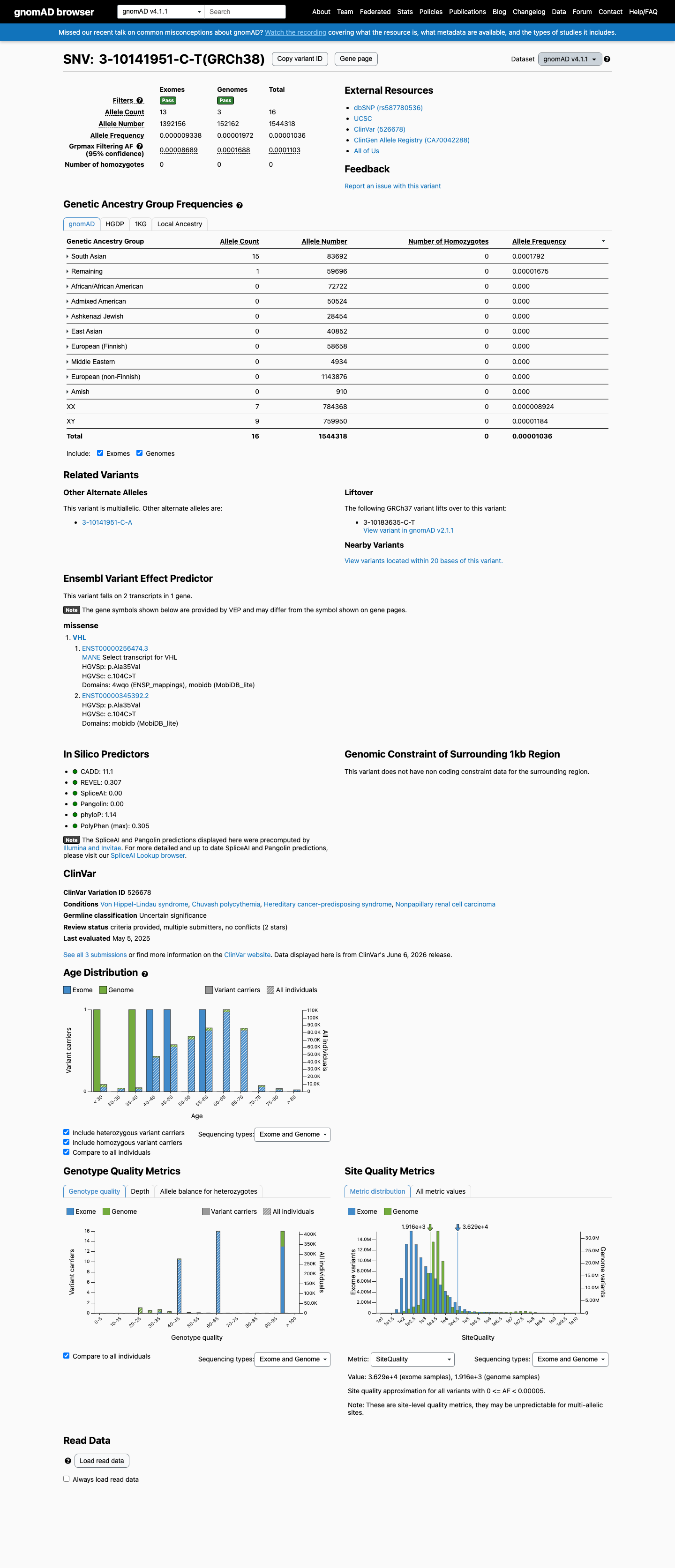
Per VHL VCEP v1.1.0, BS1 is met at Strong strength. The gnomAD v4.1 GroupMax Filtering Allele Frequency is 0.0001103 (0.01103%), which exceeds the VCEP BS1 cutoff of ≥0.0000156 (0.00156%). The variant is present at 16 alleles in gnomAD v4.1 (AF = 1.04e-5) with the highest subpopulation frequency in the South Asian population (AF = 0.000179, 15/83,692 alleles). This frequency is higher than expected for von Hippel-Lindau disease.
gnomAD v4.1: 16/1544318 alleles (AF=1.04e-5)
✓
BP4
supporting
Benign
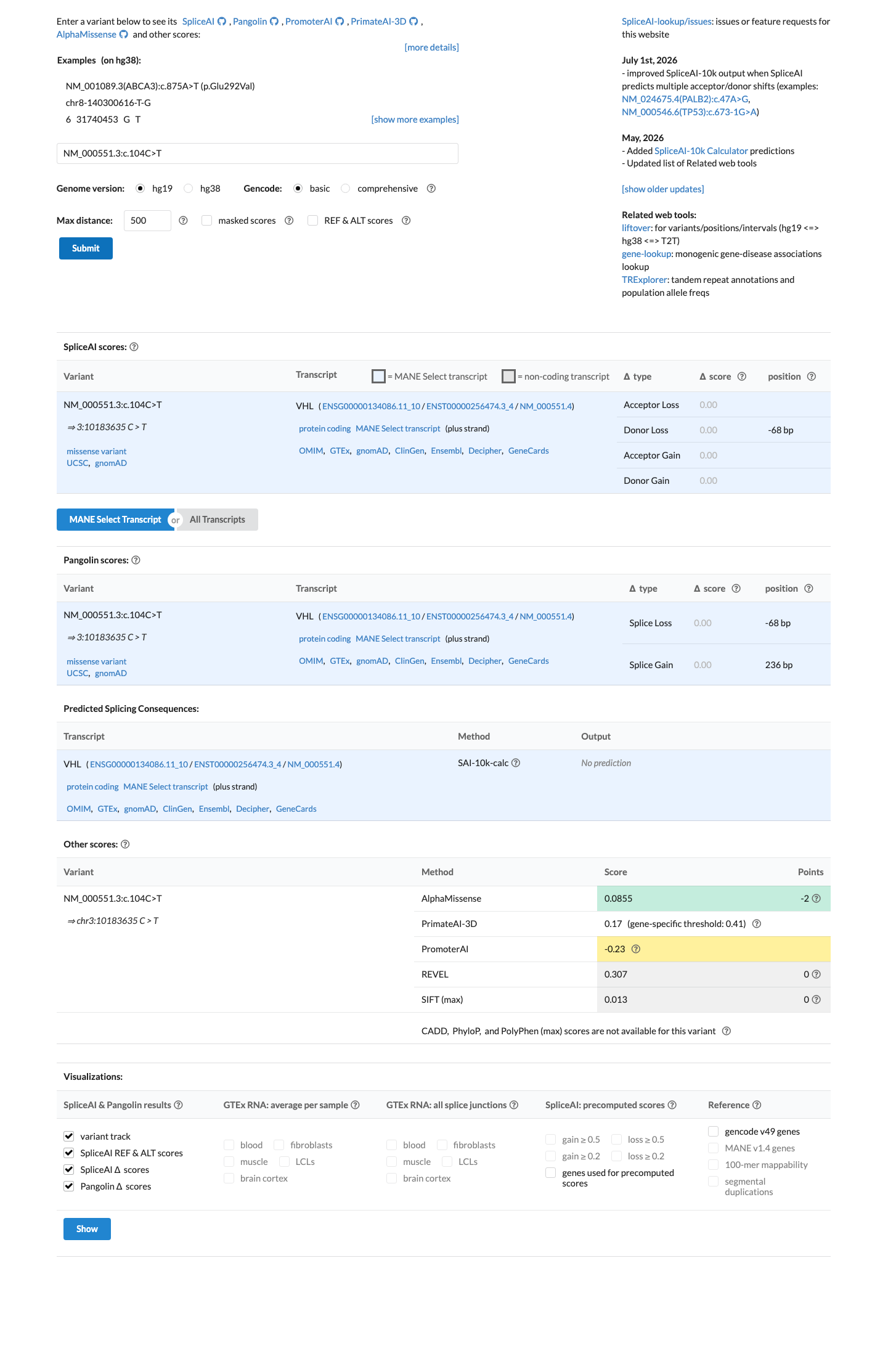
Per VHL VCEP v1.1.0, BP4 can be applied to assess lack of splicing impact. SpliceAI predicts no splicing effect (max delta score = 0.00, ≤0.1). Per VCEP guidance, SpliceAI alone may be applied when VarSeak is unable to accept the variant type. Missense predictors are explicitly excluded from BP4 per VCEP, but splicing assessment via SpliceAI is permitted. No splice effect is predicted.
SpliceAI max delta: 0.00. VCEP BP4 rule: 'BP4 can be applied to assess lack of splicing impactwith concordance of Splice AI (≤0.1).' VarSeak not assessed (splice predictor onlyVCEP allows SpliceAI alone).
Assessed · not applied
Pathogenic
PS1
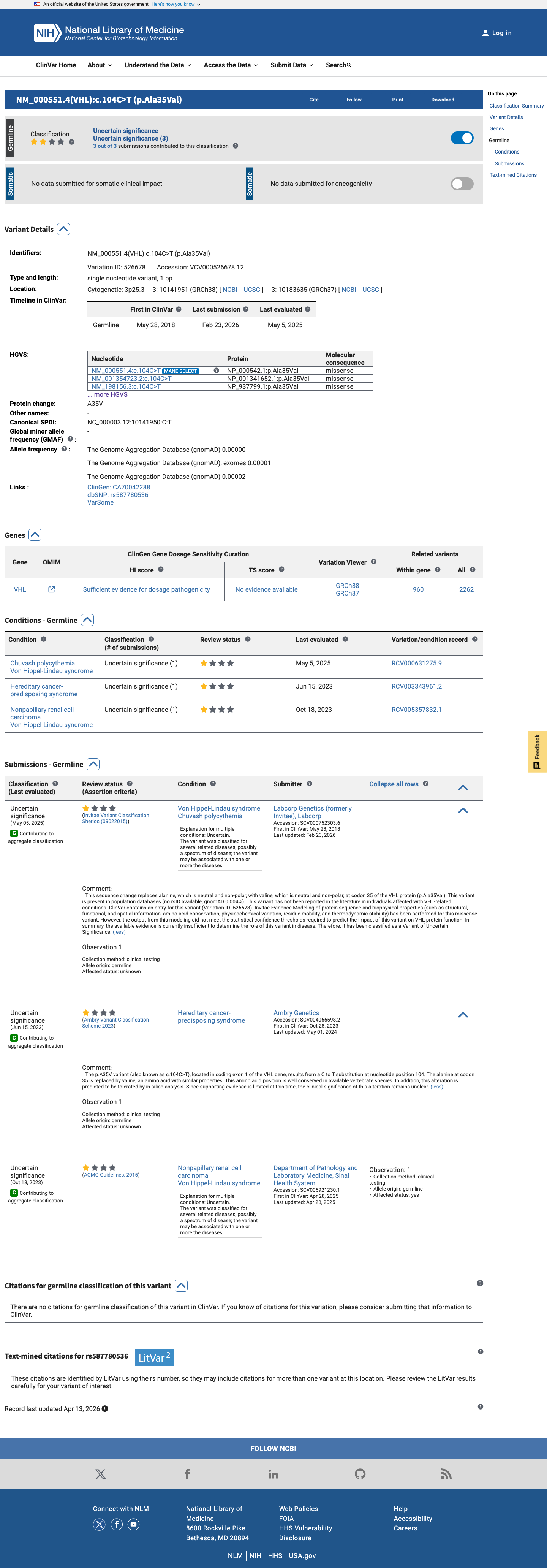
PS1 requires a different nucleotide change producing the same amino acid change (p.Ala35Val) previously classified as pathogenic by the VHL VCEP.
PS2
No de novo observations reported for NM_000551.3:c.104C>T.
PS3
No variant-specific functional data available for p.Ala35Val.
PS4
No proband count data meeting VCEP PS4 phenotype scoring criteria.
PM1
Codon 35 is located in the N-terminal region of VHL (residues 1-54), upstream of both the p19 initiation site (Met54) and the critical functional domains.
PM2
Per VHL VCEP v1.1.0, PM2_Supporting requires absence from gnomAD or GroupMax Filtering Allele Frequency (FAF) ≤ 0.00000156 (0.000156%) in gnomAD v4.
PM5
PM5 requires a different pathogenic missense variant at the same residue (Ala35) classified by the VHL VCEP.
PM6
PM6 requires assumed de novo occurrence without confirmation of maternity and paternity.
PP1
PP1 requires co-segregation data with disease in multiple affected family members.
PP3
Per VHL VCEP v1.1.0, PP3 for missense variants requires REVEL score ≥ 0.664.
Benign
BA1
Per VHL VCEP v1.1.0, BA1 requires GroupMax Filtering Allele Frequency ≥ 0.000156 (0.0156%) in gnomAD v4.
BS2
VHL VCEP BS2 requires at least 3 individuals ≥65 years old harboring the variant, unaffected by VHL-related cancers.
BS3
VHL VCEP BS3 requires well-established functional studies showing no damaging effect (HIF1/2a degradation preserved, VBC complex stability unaffected, ECM/fibronectin binding unaffected).
BS4
VHL VCEP BS4 requires lack of segregation in affected family members who fulfill Danish Criteria for VHL.
BP2
VHL VCEP BP2 requires observation in trans with a pathogenic VHL variant (phase confirmed, no congenital polycythemia), or in homozygous state without disease, or in cis with three or more pathogenic VHL variants.
BP5
VHL VCEP BP5 requires two or more co-occurrences with pathogenic variants in a different gene that fully explain the patient's phenotype.
BP7
VHL VCEP BP7 is restricted to synonymous (silent) or intronic variants where BP4 is met for lack of splice effect and the PhyloP score is ≤0.2.
N/A · 9
PVS1 · PM3 · PM4 · PP2 · PP4 · PP5 · BP1 · BP3 · BP6
Research & evidence
Population frequency · supports benign
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.03606e-05; MAF= 0.00104%, 16/1544318 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 0.000179229; MAF= 0.01792%, 15/83692 alleles, homozygotes = 0); grpmax FAF= 0.0001103.
v2.1
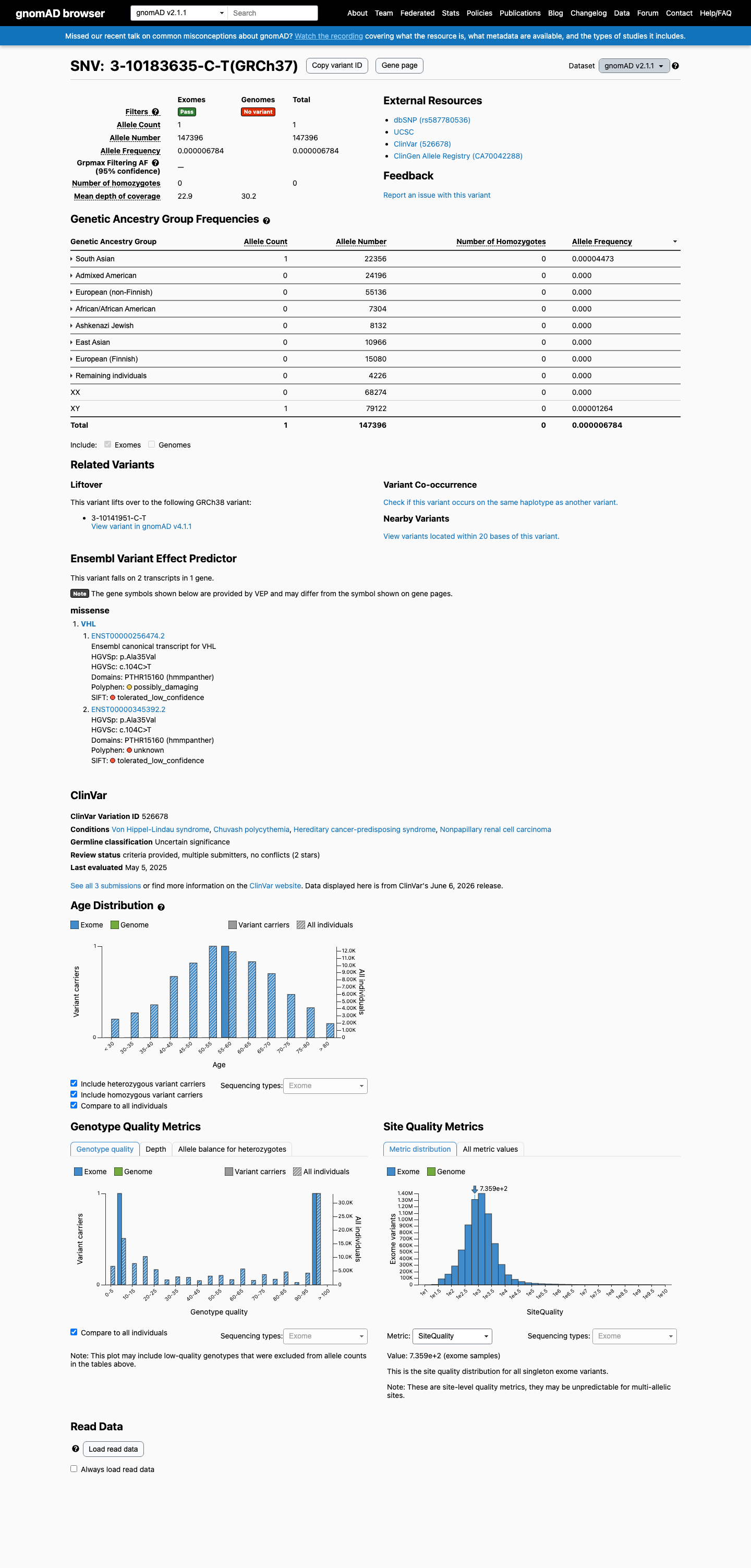
This variant is present in gnomAD v2.1 (AF= 6.78444e-06; MAF= 0.00068%, 1/147396 alleles, homozygotes = 0) and has highest observed frequency in the South Asian population (AF= 4.47307e-05; MAF= 0.00447%, 1/22356 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.001%
· 16 / 1,544,318
0 hom · FAF 0.011%
0 hom · FAF 0.011%
South Asian 15 / 83,692 |
0.018% |
Remaining individuals 1 / 59,696 |
0.0017% |
+ 8 not observed (Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
0.00068%
· 1 / 147,396
0 hom
0 hom
South Asian 1 / 22,356 |
0.0045% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
This variant has been reported in ClinVar as Uncertain significance (3 clinical laboratories). (ClinVarID = 526678)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.307. BayesDel score = -0.264275.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. VHL, an E3 ubiquitin ligase, is frequently mutated in renal cell carcinomas.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
9Sources
Triaged references · 6 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
15604628 ↗
Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors.
CLINVAR
25356965 ↗
ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing.
CLINVAR