NM_000719.7:c.5779C>G (p.His1927Asp) in CACNA1C is a missense variant in exon 45 classified as Uncertain significance in ClinVar (VariationID 1481507, 2 clinical laboratories).1 This variant is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (AF=0.00025%, 4/1,606,858 alleles, 0 homozygotes; grpmax FAF=8e-7), well below the 0.1% PM2 threshold for population rarity (PM2_Supporting).2 Multiple in silico tools suggest no deleterious impact: SpliceAI predicts no splicing effect (max delta 0.02) and BayesDel score (0.079) falls in the benign range; REVEL score (0.378) is intermediate and below established pathogenic thresholds (BP4_Supporting).3 No functional studies, case-control data, segregation analysis, de novo observations, or same-residue pathogenic comparators were identified in the reviewed literature or ClinVar submissions.4 Four publications cited in ClinVar were reviewed: a GeneReviews overview (PMID:20301308), an expert consensus statement on inherited arrhythmias (PMID:23994779), the ACMG SF v3.1 policy statement (PMID:35802134), and the Sherloc variant classification framework (PMID:28492532). None of these publications mention or study this specific variant.5
CACNA1C
Final classification
VUS
CACNA1C c.5779C>G · p.His1927Asp
CACNA1C
NM_000719.7:c.5779C>G (p.His1927Asp) in CACNA1C is a missense variant in exon 45 classified as Uncertain significance in ClinVar (VariationID 1481507, 2 clinical laboratories).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign; combination = 1 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PM2
BP4
VUS
CACNA1C c.5779C>G
PM2 + BP4
→
VUS
Gene diagram
· NM_000719.7 · variants mapped to exon structure
CACNA1C
NM_000719.7
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 20 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (AF=0.00025%, 4/1,606,858 alleles, 0 homozygotes; grpmax FAF=8e-7), well below the 0.1% PM2 threshold. Also absent from gnomAD-Canada v1.0.
gnomAD v2.1: absentgnomAD v4.1: AF=2.49e-6 (4/1606
✓
BP4
supporting
Benign
Multiple in silico tools suggest no deleterious impact: SpliceAI predicts no splicing effect (max delta 0.02), and BayesDel score (0.079) is in the benign range below standard pathogenic thresholds. REVEL score (0.378) is intermediate and below the established pathogenic threshold (>0.5).
SpliceAI max delta=0.02 (no splicing impact)BayesDel=0.079 (benign range)REVEL=0.378 (intermediate
Assessed · not applied
Pathogenic
PS1
No evidence that a different nucleotide substitution at codon His1927 has been classified as pathogenic.
PS2
No confirmed de novo occurrence reported for this variant in any publication or ClinVar submission.
PS3
No well-established functional studies demonstrating a deleterious effect for this variant have been identified.
PS4
No case-control studies demonstrate statistically significant enrichment of this variant in affected individuals.
PM1
Residue His1927 is not located within a statistically significant mutational hotspot in CACNA1C.
PM6
No de novo observations (with or without confirmed paternity/maternity) have been reported for this variant.
PP1
No co-segregation data available for this variant in affected families.
PP2
Insufficient constraint data to establish that CACNA1C has a low rate of benign missense variation.
PP3
In silico predictions do not support a deleterious effect: REVEL score 0.378 is below the typical pathogenic threshold (>0.5), BayesDel score 0.079 is in the benign range, and SpliceAI predicts no splicing impact (max delta 0.02).
PP4
No phenotype or family history data are available for individuals carrying this variant.
PP5
No reputable source reports this variant as pathogenic.
Benign
BA1
Allele frequency in gnomAD v4.1 is 0.00025% (4/1,606,858), far below the 1% BA1 threshold.
BS1
Allele frequency in gnomAD v4.1 is 0.00025% (4/1,606,858), below the 0.3% BS1 threshold for a dominant disorder.
BS2
No evidence that this variant has been observed in healthy adult controls with full penetrance data.
BS3
No well-established functional studies demonstrate no deleterious effect for this variant.
BS4
No segregation data available to demonstrate lack of co-segregation with disease.
BP1
While CACNA1C loss-of-function is a supported disease mechanism, missense variants are also a well-established pathogenic mechanism in this gene (e.g., p.Gly406Arg causing Timothy syndrome).
BP2
No evidence of this variant observed in trans with a pathogenic variant in a dominant disorder.
BP5
No evidence of an alternate molecular basis for disease in individuals carrying this variant.
BP6
No reputable source classifies this variant as benign.
N/A · 3
PVS1 · PM5 · BP7
Research & evidence
Population frequency
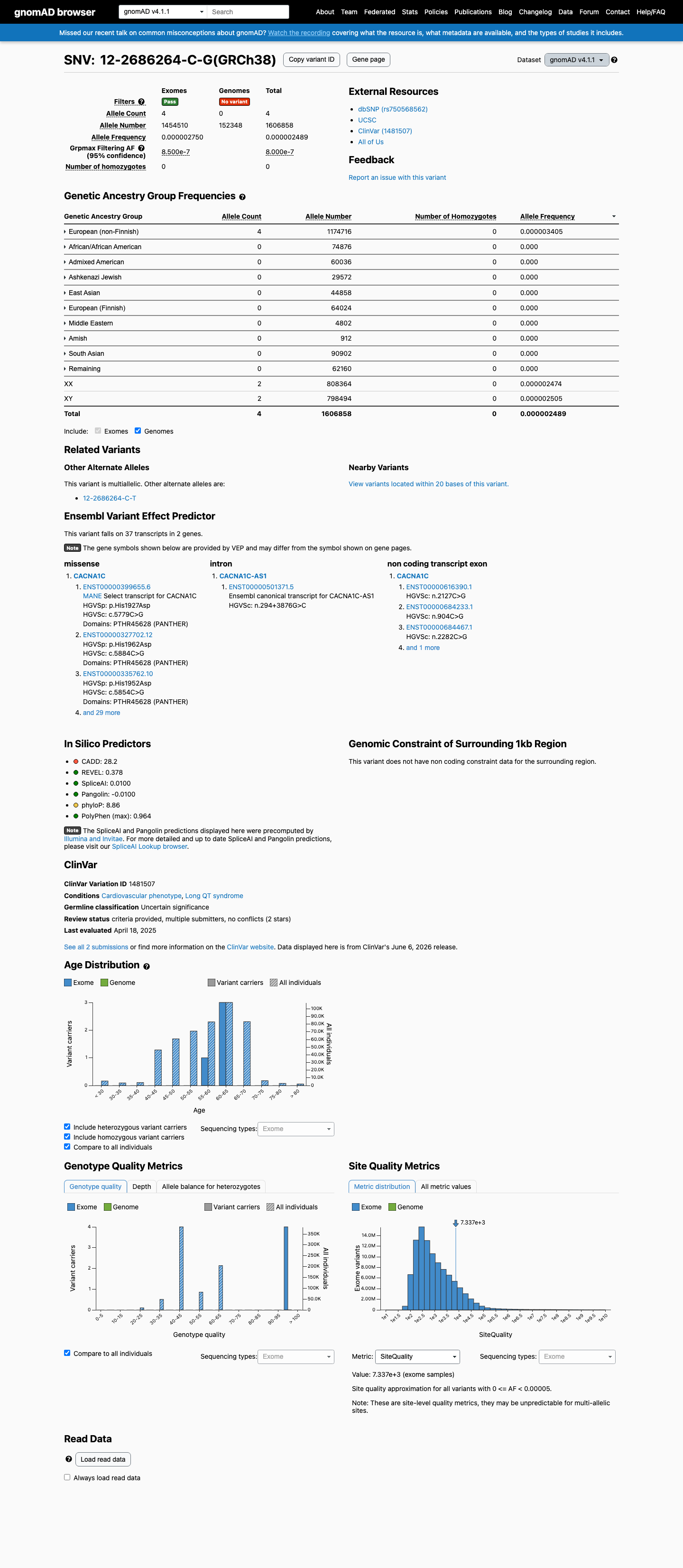
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 2.48933e-06; MAF= 0.00025%, 4/1606858 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.40508e-06; MAF= 0.00034%, 4/1174716 alleles, homozygotes = 0); grpmax FAF= 8e-07.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00025%
· 4 / 1,606,858
0 hom · FAF 8e-05%
0 hom · FAF 8e-05%
European (non-Finnish) 4 / 1,174,716 |
0.00034% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
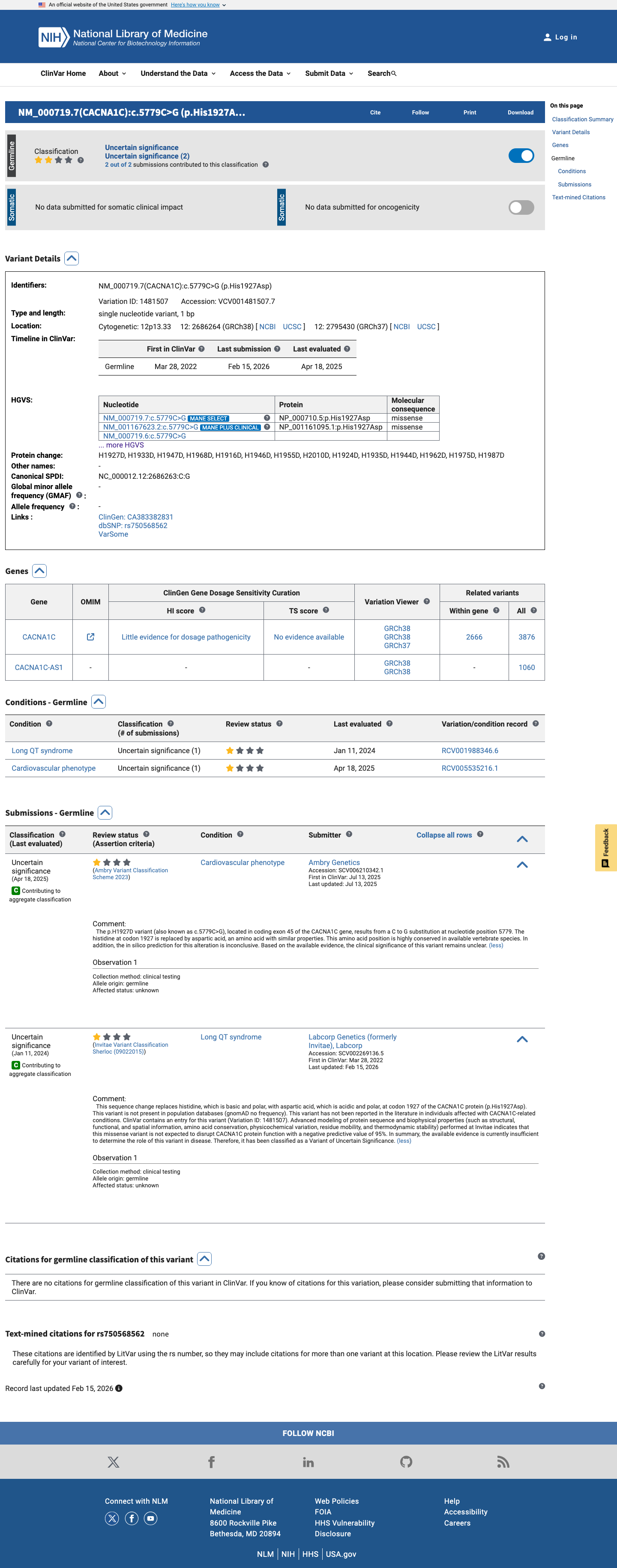
ClinVar
This variant has been reported in ClinVar as Uncertain significance (2 clinical laboratories). (ClinVarID = 1481507)
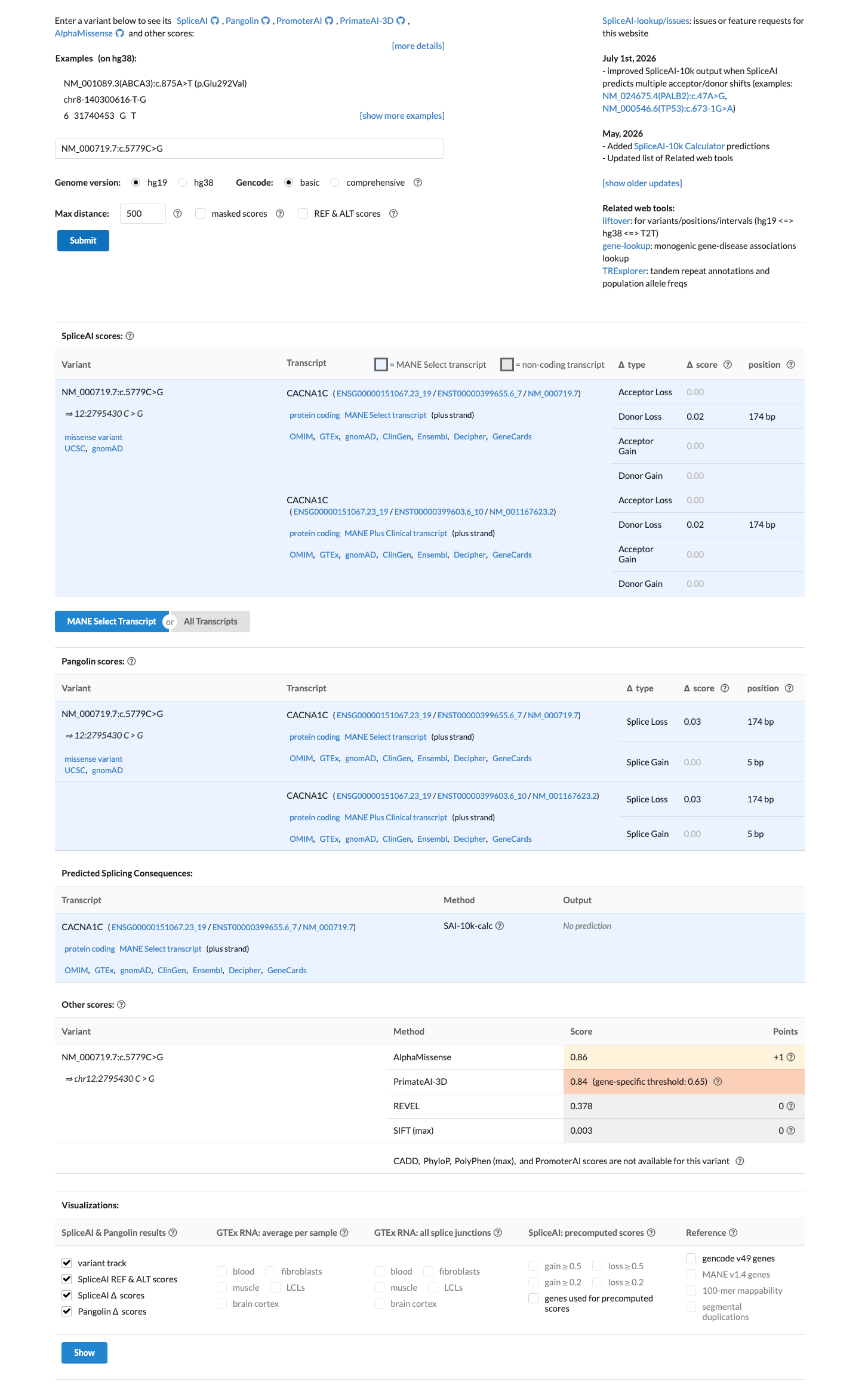
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). REVEL score = 0.378. BayesDel score = 0.0794366.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 3 PMIDs not cited in assessment
23994779 ↗
Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes.
CLINVAR
35802134 ↗
ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR