NM_001023.4:c.24A>G (p.Lys8=) is a synonymous variant in exon 2 of RPS20 with no predicted effect on splicing (SpliceAI max delta = 0.00).1 This variant has been classified as Likely benign in ClinVar by a single clinical laboratory (Ambry Genetics, criteria provided; variation ID 2488528).2 The variant is present at extremely low frequency in population databases (gnomAD v2.1: 1/249,106 alleles, AF = 0.0004%; v4.1: 1/1,613,194 alleles, AF = 0.00006%), but rarity alone does not confer pathogenicity for a synonymous change with no functional consequence.3 No functional studies, case-control data, de novo reports, segregation data, or publications mentioning this specific variant were identified. Three supporting benign criteria are met: BP4 (computational evidence predicts no impact), BP6 (reputable source reports as benign), and BP7 (synonymous variant with no splice effect). This is consistent with a Likely benign classification per ACMG/AMP 2015 guidelines.4
RPS20
Final classification
Likely Benign
RPS20 c.24A>G · p.Lys8=
RPS20
NM_001023.4:c.24A>G (p.Lys8=) is a synonymous variant in exon 2 of RPS20 with no predicted effect on splicing (SpliceAI max delta = 0.00).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, BP4 supporting benign, BP6 supporting benign, BP7 supporting benign; combination = 1 supporting + 3 supporting benign, which maps to Likely Benign.
Classification rationale
PM2
BP4BP6BP7
Likely Benign
RPS20 c.24A>G
PM2 + BP4 + BP6 + BP7
→
Likely Benign
Gene diagram
· NM_001023.4 · variants mapped to exon structure
RPS20
NM_001023.4
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 17 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is present at an extremely low frequency in population databases: gnomAD v2.1 AF = 4.01e-6 (1/249,106 alleles) and v4.1 AF = 6.20e-7 (1/1,613,194 alleles), well below the 0.1% PM2 threshold. However, this is a synonymous variant with no other evidence of pathogenicity, limiting the weight of rarity alone.
gnomAD v2.1: 1 allele in 249106 (AF = 0.0004%)gnomAD v4.1: 1 allele in 1
✓
BP4
supporting
Benign
SpliceAI predicts no splice impact for this variant (max delta score = 0.00). The variant is synonymous (p.Lys8=) and is predicted not to alter splicing or protein sequence. Multiple lines of computational evidence are consistent with no functional impact.
SpliceAI max delta = 0.00 (no donor gaindonor lossacceptor gain
✓
BP6
supporting
Benign
This variant has been reported in ClinVar as Likely benign by Ambry Genetics, a clinical testing laboratory, with criteria provided. Although from a single submitter, the classification supports a benign interpretation in the absence of any pathogenic evidence.
ClinVar variation ID 2488528: Likely benign (Ambry Geneticscriteria providedsingle submitter)
✓
BP7
supporting
Benign
This is a synonymous variant (p.Lys8=) with no predicted splice impact (SpliceAI max delta = 0.00). BP7 is met for a silent variant with no evidence of altered splicing.
Synonymous substitution: NP_001014.1:p.(Lys8=)SpliceAI max delta = 0.00 — no predicted splice alterationVariant is not in a highly conserved nucleotide position with evidence of splicing effect
Assessed · not applied
Pathogenic
PS1
No alternative nucleotide change at c.24 resulting in the same amino acid (p.Lys8=) has been reported as pathogenic.
PS2
No de novo data are available.
PS3
No functional studies have been identified for this variant.
PS4
No case-control or cohort data comparing variant prevalence in affected versus unaffected individuals are available.
PM1
This variant is not located in a statistically significant mutational hotspot (cancerhotspots.org negative), and as a synonymous variant (p.Lys8=) it does not alter any amino acid residue in a critical functional domain.
PM6
No de novo data are available.
PP1
No co-segregation data are available.
PP3
No in silico predictors suggest a deleterious effect.
PP4
No patient phenotype or clinical data are available to assess whether the presentation is highly specific for an RPS20-related disorder.
PP5
ClinVar classifies this variant as Likely benign, not pathogenic.
Benign
BA1
The maximum allele frequency in gnomAD is 4.01e-6 (0.0004%), far below the 1% BA1 threshold.
BS1
The maximum allele frequency in gnomAD is 4.01e-6 (0.0004%), well below the 0.3% BS1 threshold.
BS2
No data are available regarding observation of this variant in healthy adults with full penetrance expected at their age.
BS3
No well-established functional studies have evaluated this variant for a deleterious effect.
BS4
No segregation data are available to assess lack of co-segregation with disease.
BP2
No data are available regarding observation of this variant in trans with a known pathogenic variant.
BP5
No data are available regarding an alternative molecular basis for disease in a case harboring this variant.
N/A · 4
PVS1 · PM5 · PP2 · BP1
Research & evidence
Population frequency · supports benign
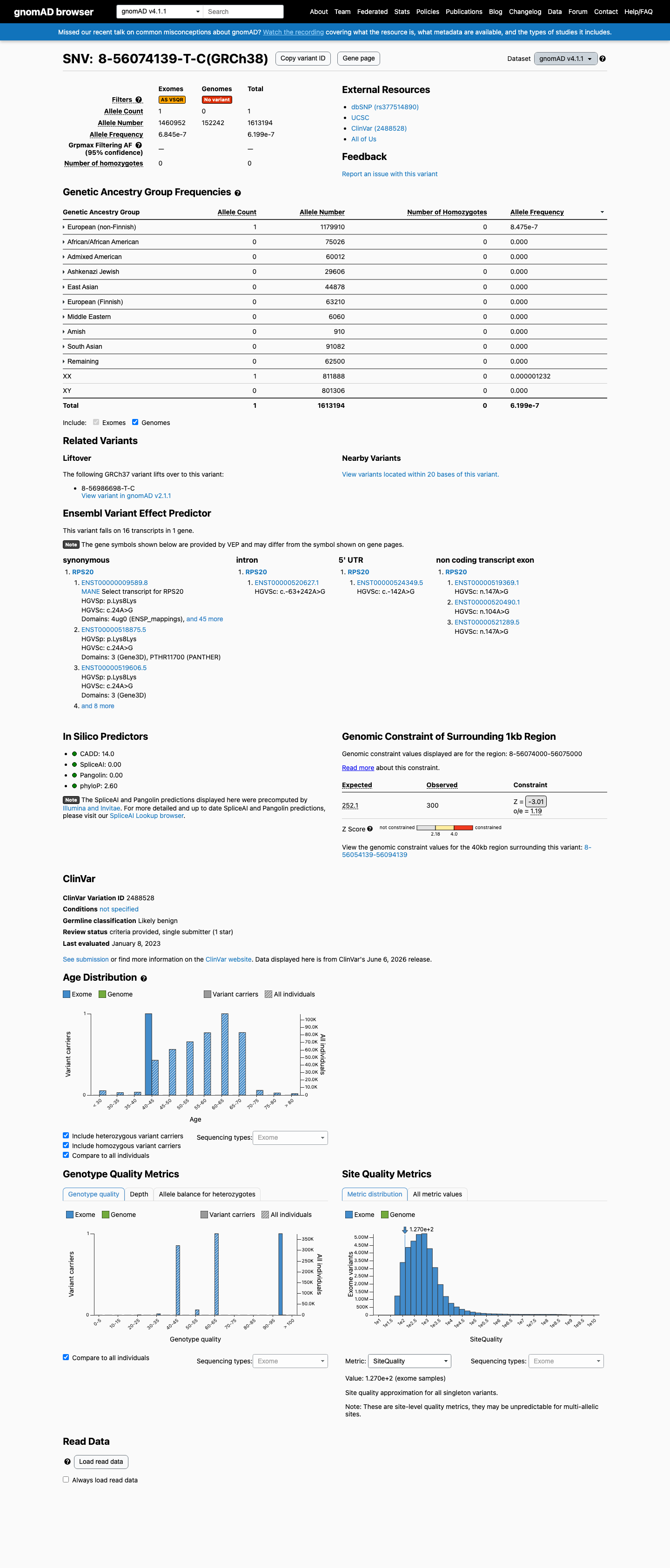
gnomAD v4.1
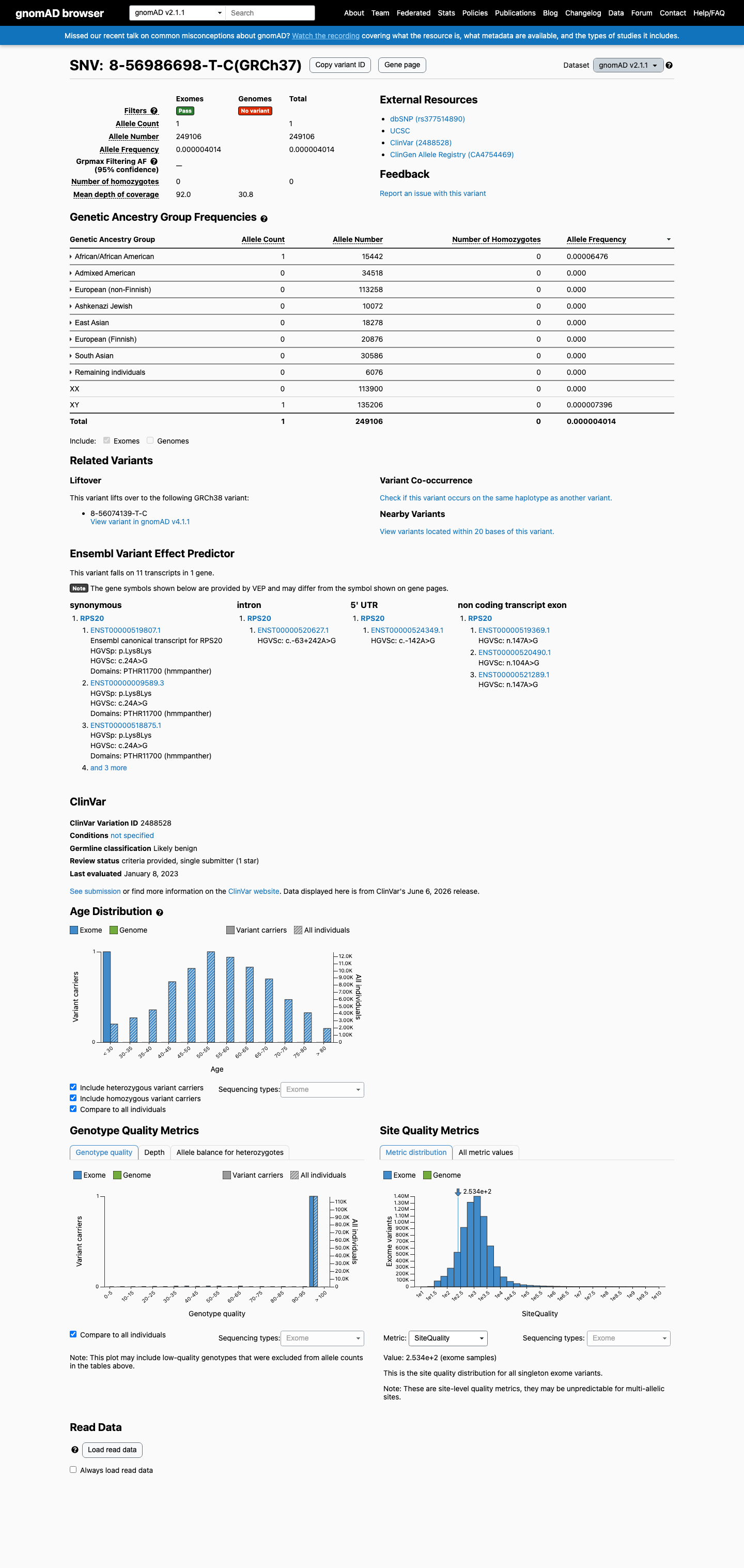
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 6.19888e-07; MAF= 0.00006%, 1/1613194 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.47522e-07; MAF= 0.00008%, 1/1179910 alleles, homozygotes = 0).
v2.1
This variant is present in gnomAD v2.1 (AF= 4.01436e-06; MAF= 0.00040%, 1/249106 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 6.47585e-05; MAF= 0.00648%, 1/15442 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,613,194
0 hom
0 hom
European (non-Finnish) 1 / 1,179,910 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 249,106
0 hom
0 hom
African/African American 1 / 15,442 |
0.0065% |
+ 7 not observed (Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
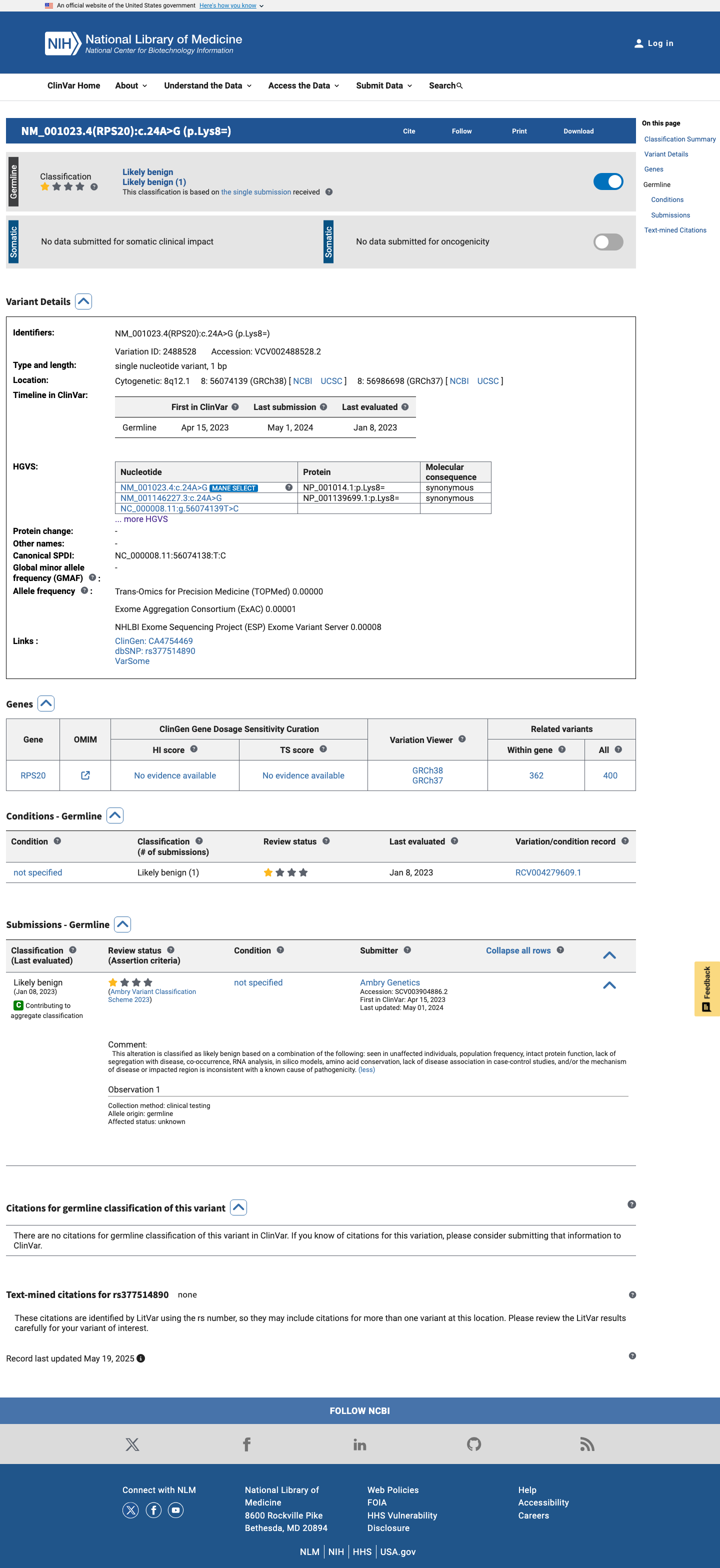
ClinVar
This variant has been reported in ClinVar as Likely benign (1 clinical laboratory). (ClinVarID = 2488528)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00).
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links