Classification rationale
PM2
BP4
VUS
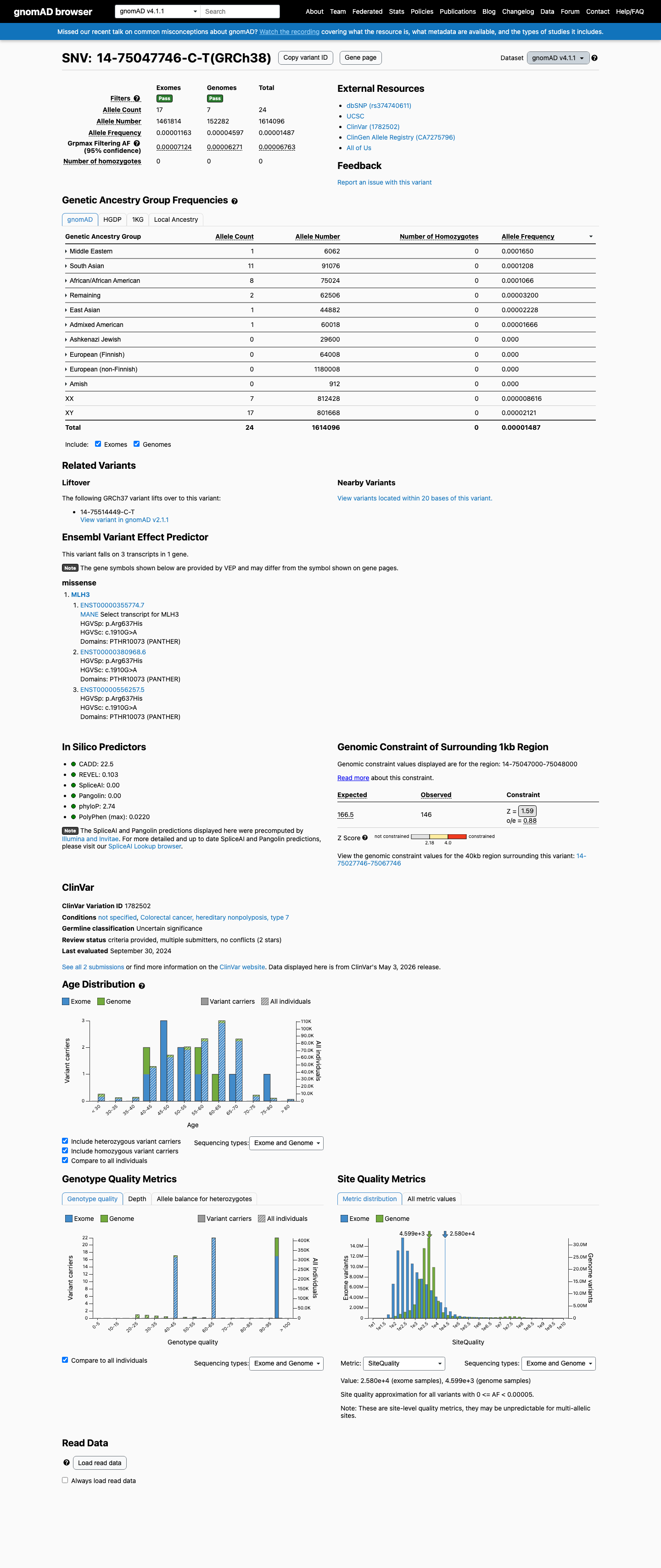
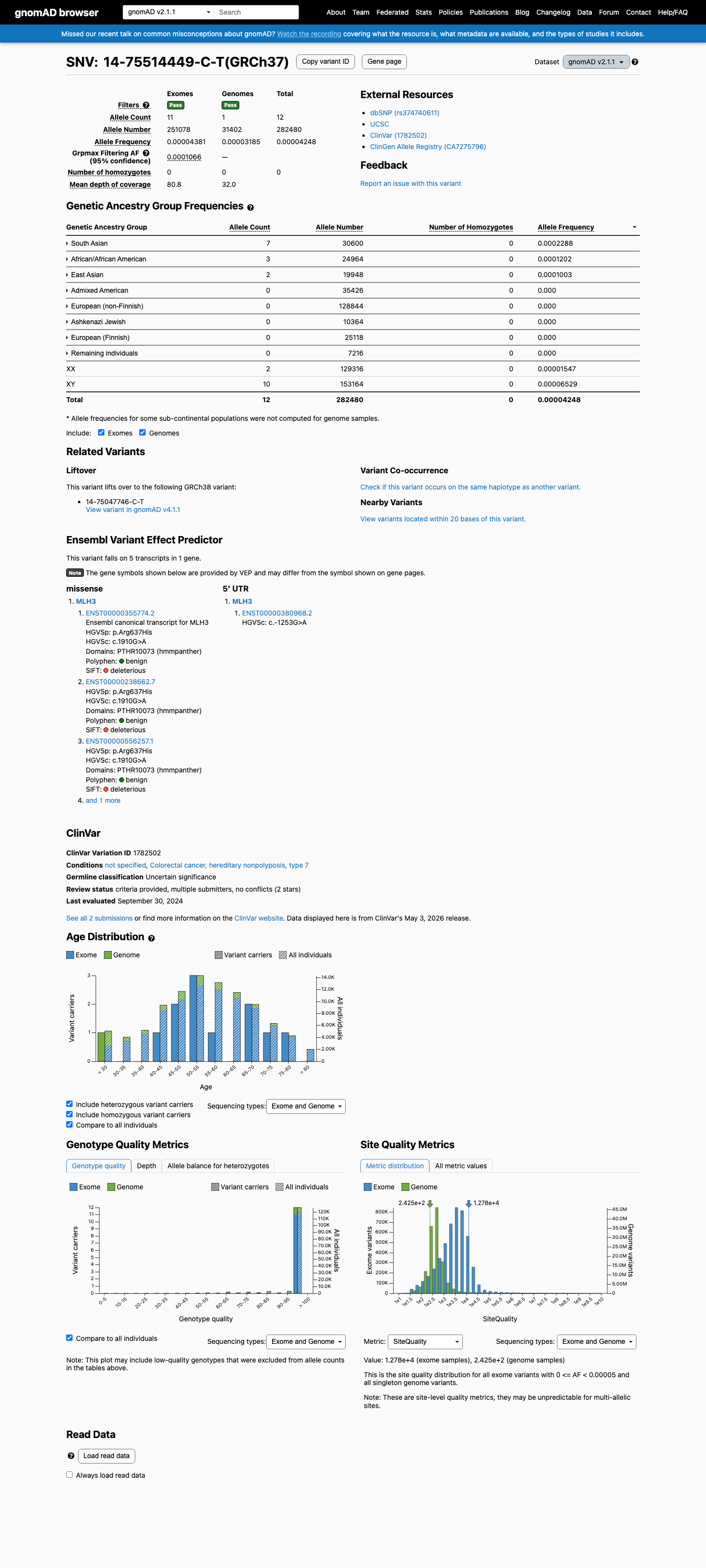
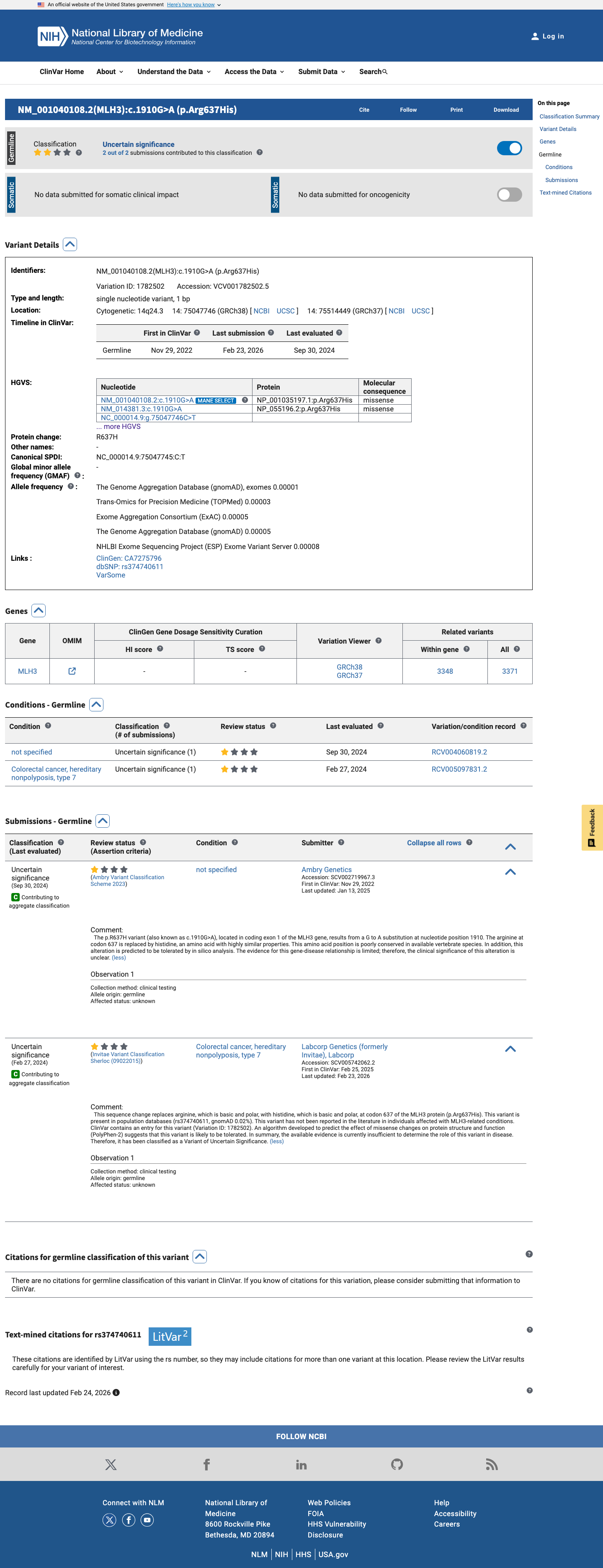
MLH3 c.1910G>A
The MLH3 c.1910G>A (p.Arg637His, p.R637H) variant has been reported in ClinVar, where current submissions classify it as a variant of uncertain significance.1 This variant is rare in population databases, with a total allele frequency of 0.00425% in gnomAD v2.1 and 0.00149% in gnomAD v4.1, and it is absent from gnomAD-Canada v1.0.2 Available computational evidence does not support a damaging effect: REVEL is 0.103, BayesDel is -0.485573, and SpliceAI predicts no significant splice impact with a maximum delta score of 0.00.3
PM2 + BP4
→
VUS