Classification rationale
BA1BS1BP4
Benign
MLH3 c.3488G>A
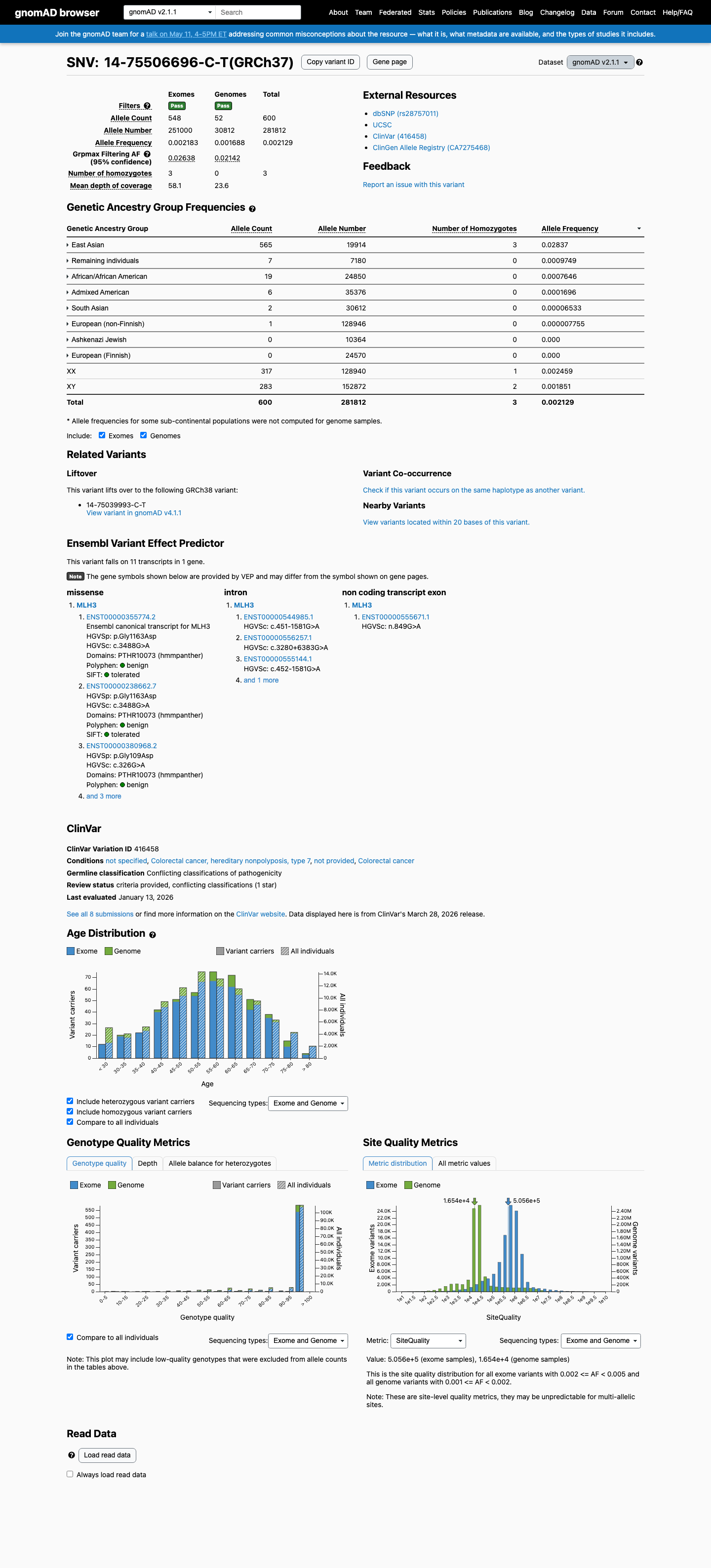
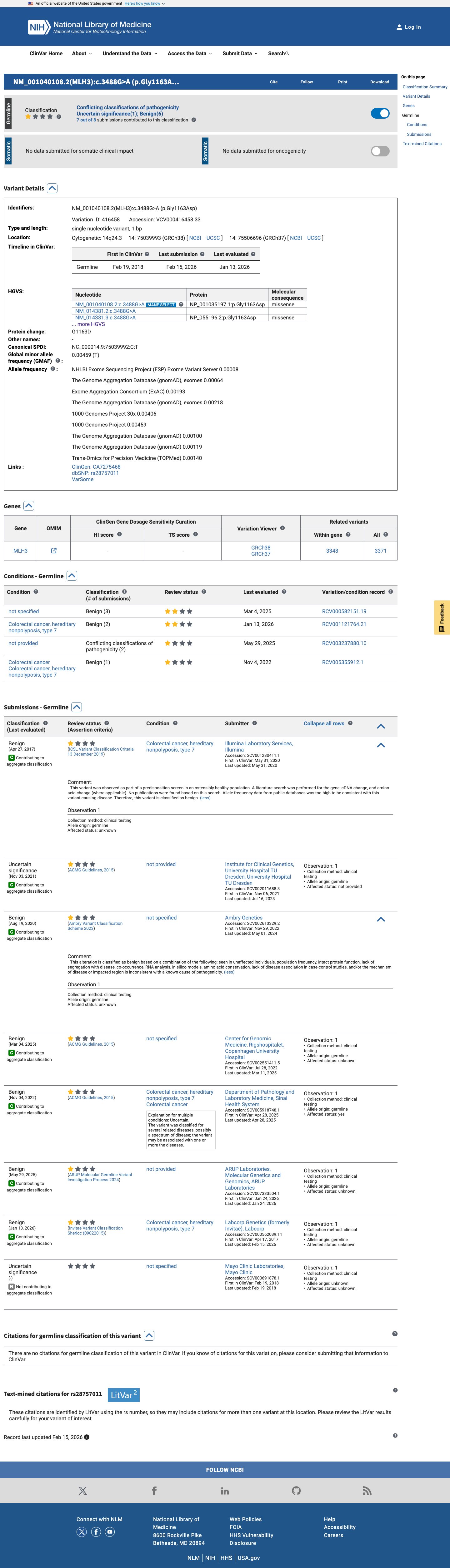
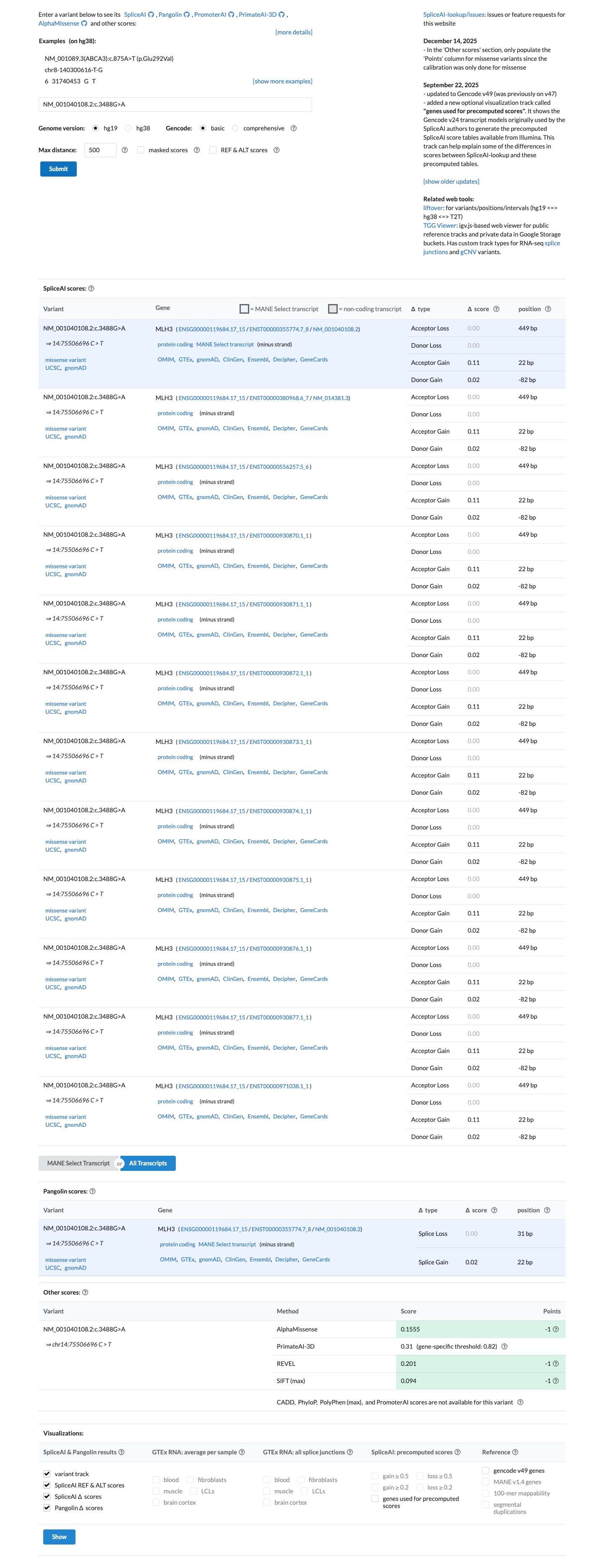
The MLH3 c.3488G>A (p.Gly1163Asp; p.G1163D) variant has been reported in ClinVar, where most submissions classify it as benign (6 laboratories) and a minority classify it as uncertain significance (2 laboratories).1 This variant is common in population databases, with East Asian allele frequencies of 2.83720% in gnomAD v2.1 and 2.24070% in gnomAD v4.1, exceeding the default BA1 threshold of 1% and BS1 threshold of 0.3%.2 Computational evidence argues against a deleterious effect: SpliceAI predicts no significant splice impact with a max delta score of 0.00, REVEL is 0.113, and BayesDel is -0.258625.3
BA1 + BS1 + BP4
→
Benign