NM_001127208.2:c.4354C>T (p.Arg1452Ter) is a nonsense variant in exon 10 of 11 of TET2, predicted to result in nonsense-mediated decay and loss of protein function.1 TET2 germline loss-of-function mutations are associated with an ALPS-like immune dysregulation syndrome and predisposition to hematologic malignancy, with heterozygous LoF variants reported to cause disease in multiple families.2 This variant has been observed in gnomAD at extremely low frequency (v2.1: 2/156592 alleles, AF=0.00128%; v4.1: 15/1551500 alleles, AF=0.00097%) with no homozygotes, and is absent from gnomAD-Canada.3 The variant truncates the protein at residue 1452 within the catalytic domain (residues 1129-1936), removing critical functional regions including the DSBH core, Fe(II)-chelating site (H1881), 2OG-interacting residues (R1896), and DNA-interacting loops.4 This variant has been observed in COSMIC as a somatic mutation (27 entries) and reported in ClinVar with conflicting classifications (one VUS, one Pathogenic, single submitters with no expert panel review).5 No variant-specific functional studies, de novo reports, segregation data, or case-control studies were identified for c.4354C>T in the reviewed literature.6
TET2
Final classification
Likely Pathogenic
TET2 c.4354C>T · p.Arg1452Ter
TET2
NM_001127208.2:c.4354C>T (p.Arg1452Ter) is a nonsense variant in exon 10 of 11 of TET2, predicted to result in nonsense-mediated decay and loss of protein function.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PVS1 strong, PM1 moderate, PM2 supporting; combination = 1 strong + 1 moderate + 1 supporting, which maps to Likely Pathogenic.
Classification rationale
PVS1PM1PM2
Likely Pathogenic
TET2 c.4354C>T
PVS1 + PM1 + PM2
→
Likely Pathogenic
1
pvs1_variant_assessmentpvs1_generic_framework ↗
2
PMID:36066697 ↗pvs1_gene_context
Gene diagram
· NM_001127208.2 · variants mapped to exon structure
TET2
NM_001127208.2
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 17 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
strong
Pathogenic
This variant is a nonsense change (c.4354C>T, p.Arg1452Ter) predicted to result in premature termination and nonsense-mediated decay (NMD) in a gene where loss of function is an established germline disease mechanism. The variant is located in exon 10 of 11 and is not predicted to escape NMD. TET2 germline loss-of-function mutations are associated with an ALPS-like immune dysregulation syndrome with predisposition to hematologic malignancy (PMID:36066697, PMID:40031954). Under PMC6185798, nonsense variants in genes with established LoF disease mechanism qualify for PVS1 at full strength when not in the terminal exon.
Nonsense variant (p.Arg1452Ter) in exon 10 of 11predicted to trigger NMDTET2 germline loss-of-function established as disease mechanism for ALPS-like syndrome and lymphoma predisposition
✓
PM1
moderate
Pathogenic
The variant truncates the TET2 protein at position 1452, which lies within the catalytic domain (residues 1129-1936 as defined by PMID:24315485). The truncation removes the DSBH core and C-terminal catalytic machinery including Fe(II)-chelating residues (H1881), 2OG-interacting residues (R1896), and DNA-interacting loops. The catalytic domain is a well-characterized functional domain critical for TET2's 5-methylcytosine dioxygenase activity. Truncating variants that remove this domain abrogate TET2 enzymatic function.
Variant truncates at residue 1452 within the catalytic domain (1129-1936)PMID:24315485 defines the catalytic domain structure and functionally critical residuesTruncation removes DSBH core
✓
PM2
supporting
Pathogenic
This variant is present in gnomAD at extremely low allele frequency: v2.1 AF=1.28e-5 (0.00128%, 2/156592 alleles), v4.1 AF=9.67e-6 (0.00097%, 15/1551500 alleles), well below the 0.1% threshold for PM2. It is absent from gnomAD-Canada. No homozygotes have been observed in any population database.
gnomAD v2.1 AF=1.28e-5 (0.00128%)2/156592 alleles0 homozygotes
Assessed · not applied
Pathogenic
PS2
No de novo report was identified for NM_001127208.2:c.4354C>T.
PS3
No variant-specific functional data or systematic range characterization covering position Arg1452 was identified in the literature.
PS4
The variant has been observed in gnomAD at extremely low frequency (v2.1: 2/156592; v4.1: 15/1551500 alleles) and reported in ClinVar with conflicting classifications (one VUS, one Pathogenic, single submitters only).
PM6
No de novo report was identified for NM_001127208.2:c.4354C>T in the reviewed literature.
PP1
No co-segregation data is available for this variant.
PP3
Computational evidence does not support a deleterious effect for this variant.
PP4
Insufficient phenotype data is available for patients carrying this variant.
PP5
No reputable source has definitively classified this variant as pathogenic.
Benign
BA1
The maximum allele frequency in gnomAD is 1.28e-5 (0.00128%) in v2.1 and 9.67e-6 (0.00097%) in v4.1, both well below the BA1 threshold of 1% (and adjusting AF).
BS1
The maximum allele frequency in gnomAD (0.00128% in v2.1, 0.00097% in v4.1) is well below the BS1 threshold of 0.3%.
BS2
No homozygous individuals have been observed in any population database (gnomAD v2.1: 0 homozygotes out of 156592 alleles; gnomAD v4.1: 0 homozygotes out of 1551500 alleles).
BS3
No functional studies demonstrate a benign effect for this variant.
BS4
No segregation data is available for this variant.
BP2
No data is available regarding observation of this variant in trans with a known pathogenic variant.
BP4
Computational evidence does not support a benign effect.
BP5
No observation of this variant in a case with an alternate molecular basis for disease is available.
BP6
This variant is not classified as Benign or Likely Benign by a reputable source.
N/A · 5
PS1 · PM5 · PP2 · BP1 · BP7
Research & evidence
Population frequency · supports pathogenic
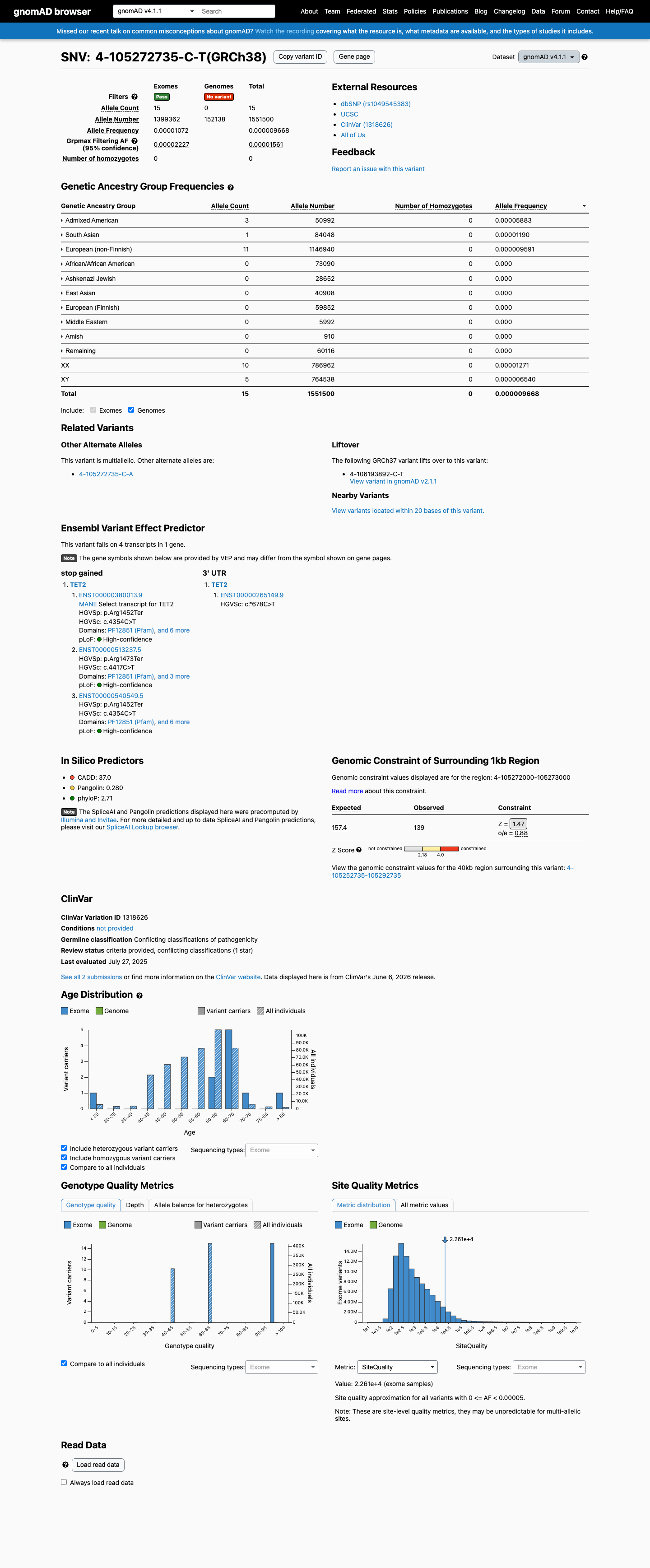
gnomAD v4.1
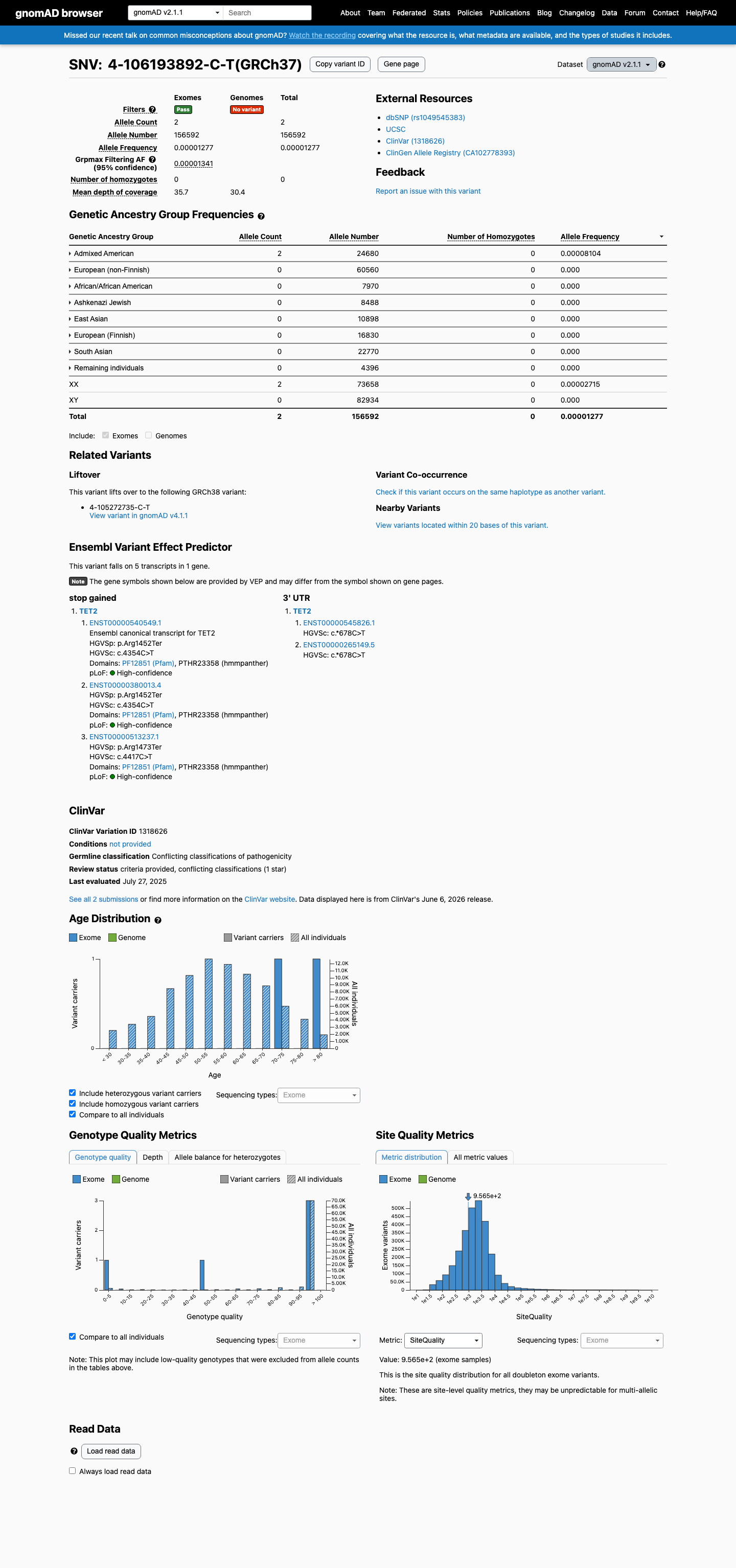
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 9.66806e-06; MAF= 0.00097%, 15/1551500 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 5.88328e-05; MAF= 0.00588%, 3/50992 alleles, homozygotes = 0); grpmax FAF= 1.561e-05.
v2.1
This variant is present in gnomAD v2.1 (AF= 1.2772e-05; MAF= 0.00128%, 2/156592 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 8.10373e-05; MAF= 0.00810%, 2/24680 alleles, homozygotes = 0); grpmax FAF= 1.341e-05.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.0001086484137331595, 2/18408 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00097%
· 15 / 1,551,500
0 hom · FAF 0.0016%
0 hom · FAF 0.0016%
Admixed American 3 / 50,992 |
0.0059% |
South Asian 1 / 84,048 |
0.0012% |
European (non-Finnish) 11 / 1,146,940 |
0.00096% |
+ 7 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0013%
· 2 / 156,592
0 hom · FAF 0.0013%
0 hom · FAF 0.0013%
Admixed American 2 / 24,680 |
0.0081% |
+ 7 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), European (non-Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
0.011%
· 2 / 18,408
0 hom · FAF 0.003%
0 hom · FAF 0.003%
European (non-Finnish) 2 / 11,728 |
0.017% |
+ 8 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, Remaining individuals, South Asian)
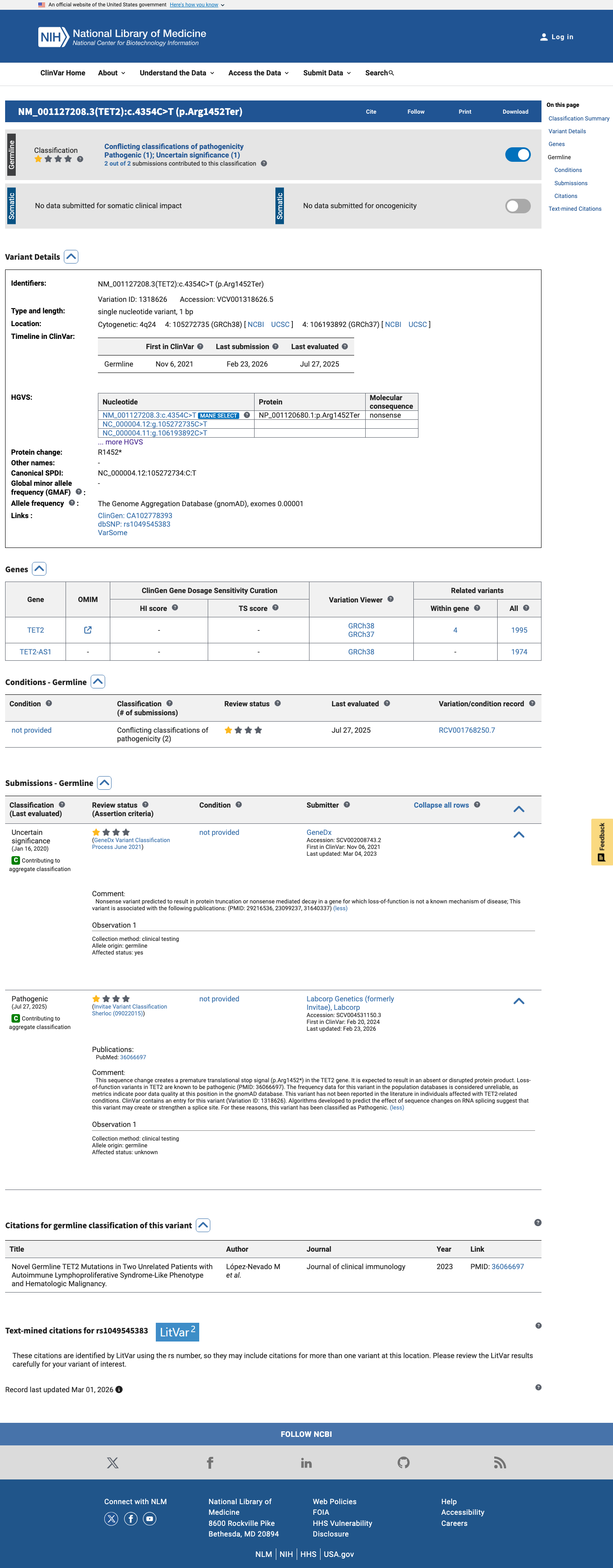
ClinVar
This variant has been reported in ClinVar as Uncertain significance (1 clinical laboratory) and as Pathogenic (1 clinical laboratory). (ClinVarID = 1318626)
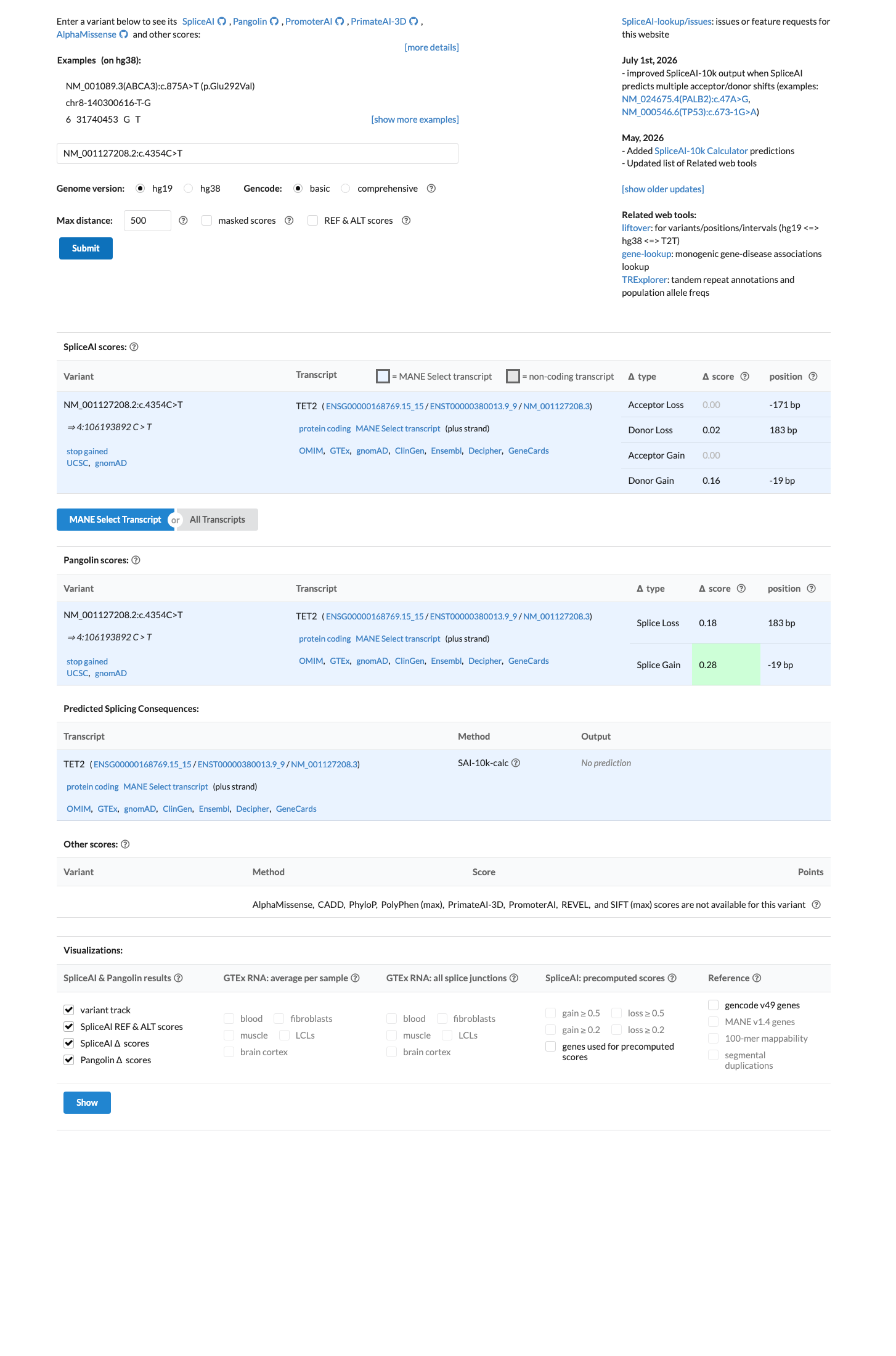
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.16). BayesDel score = 0.558747.
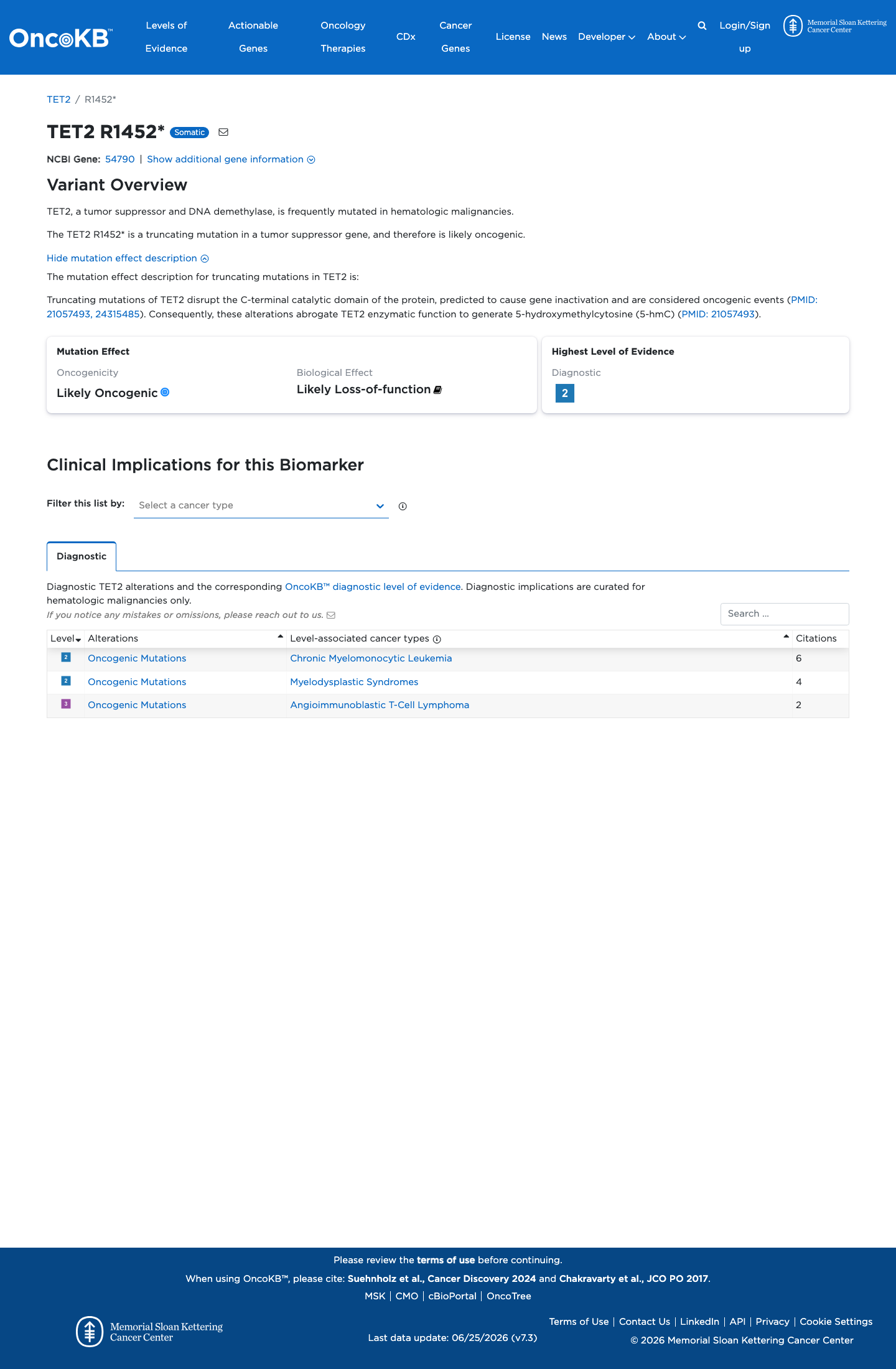
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV54398405, n = 27 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
3papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why.
Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2.
Searched
c.4354C>TR1452*Arg1452TerArg14521452
Found
Characterizes TET2 catalytic activity by expressing wild-type and mutant murine Tet2 in HEK293T cells, confirming that Tet2 converts 5-methylcytosine to 5-hydroxymethylcytosine and that mutations in Fe(II)-chelating and 2OG-interacting residues abolish enzymatic activity. Specific mutations tested include H1302Y, D1304A, H1802R, H1802Q, P1287S, W1211R, C1834D, and R1817S/M.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
Why
Variant not mentioned; cited for PM1 only as structural/functional context establishing the TET2 catalytic domain importance. Does not provide variant-specific evidence.
HEK293T cells expressing Tet2 mutants H1802R and H1802Q showed greatly diminished 5-hmC staining and no loss of 5-mC staining, consistent with participation of this residue in catalysis
Location Full text: Results section describing transient transfection and immunocytochemistry assays · Context Transient transfection of HEK293T cells with Myc-tagged murine Tet2 constructs; 5-hmC and 5-mC detection by immunocytochemistry and dot blot assays · full text
Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation.
Searched
c.4354C>TR1452*Arg1452TerArg14521452
Found
Presents the crystal structure of human TET2 catalytic domain (residues 1129-1936) bound to methylated DNA at 2.02A resolution. Identifies key catalytic residues including Fe(II)-chelating residues H1382, D1384, H1881 and NOG-interacting residues R1261, H1416, R1896, S1898. Defines domain boundaries including the low-complexity insert region (residues 1464-1843).
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
Why
Variant not mentioned; cited for PM1 as structural evidence defining the TET2 catalytic domain boundaries and functionally critical residues. Truncation at residue 1452 would remove the DSBH core and catalytic machinery.
The structure shows that two zinc fingers bring the Cys-rich and DSBH domains together to form a compact catalytic domain.
Location Results: Structure Determination and Overall Structure of TET2-DNA Complex sections · Context X-ray crystallography of human TET2 (1129-1936) with 12bp DNA and NOG; dot blot and LC-MS/MS functional assays in HEK293T cells · full text
Novel Germline TET2 Mutations in Two Unrelated Patients with Autoimmune Lymphoproliferative Syndrome-Like Phenotype and Hematologic Malignancy.
Searched
c.4354C>TR1452*Arg1452TerArg145243541452Ter
Found
Reports two unrelated ALPS-like patients with novel germline TET2 mutations: P1 (compound heterozygous for c.277G>T/p.Gly93Ter and c.1793delA/p.Asn598IlefsTer3, biallelic) and P2 (heterozygous 4q24 deletion affecting TET2, monoallelic). Both presented with T-cell lymphoma, chronic lymphoproliferation, and immune dysregulation. Functional studies confirmed absent TET2 expression and global DNA hypermethylation in the biallelic patient.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PVS1 supports · met
Why
Variant not identified. This paper establishes germline TET2 loss-of-function as a disease mechanism (supporting PVS1 gene-level eligibility) but does not report NM_001127208.2:c.4354C>T. The specific variants described (p.Gly93Ter, p.Asn598IlefsTer3, 4q24del) are different from c.4354C>T. Does not provide variant-specific evidence for PS4.
Two novel previously unreported heterozygous germline variants in TET2 (c.277G>T, p.Gly93Ter and c.1793delA, p.Asn598IlefsTer3) were detected in WES analysis and confirmed by Sanger sequencing
Location Results: Identification of Novel Germline LOF Mutations section and Table 2 · Context Whole exome sequencing, targeted NGS gene panel, Sanger sequencing, qPCR, Western blot, global DNA methylation assay, flow cytometry immunophenotyping · full text
Sources & reference links