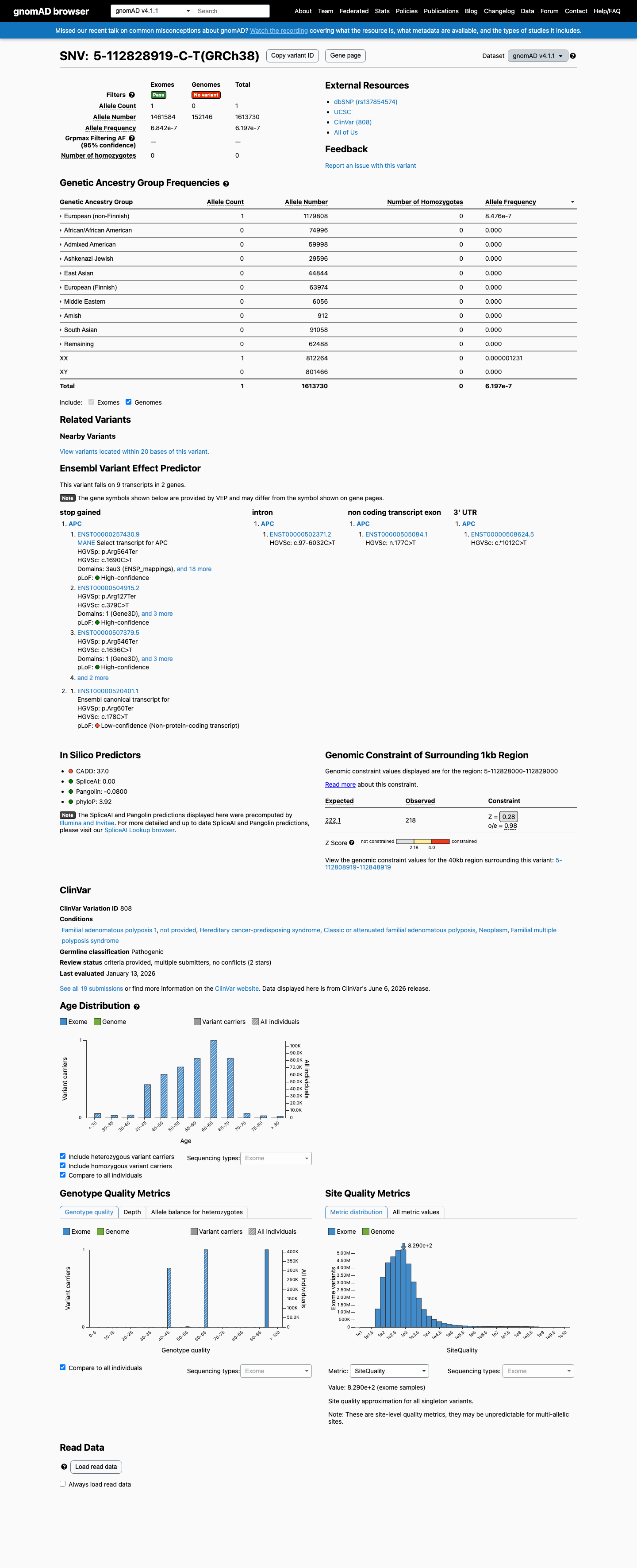
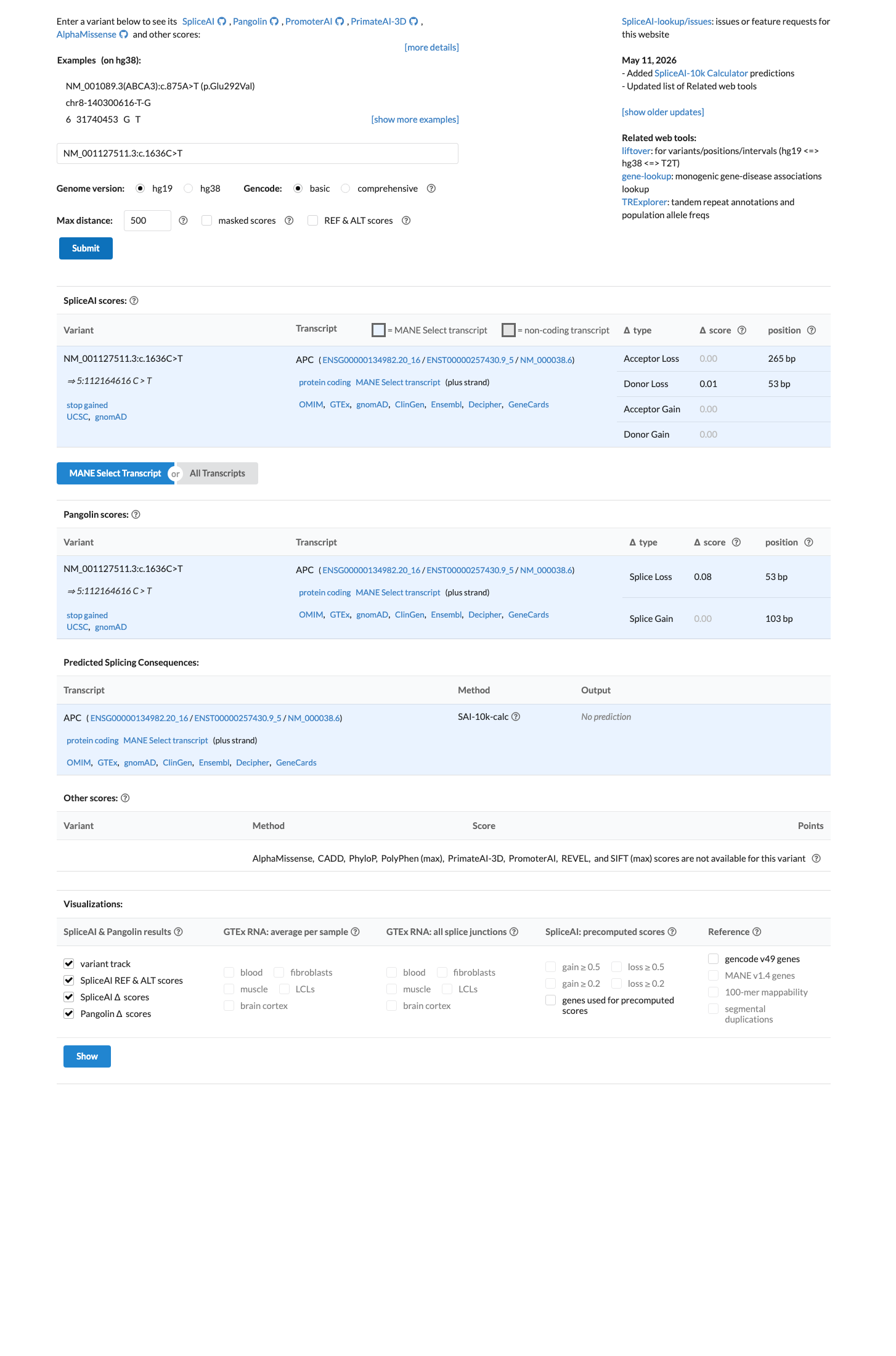
NM_001127511.3:c.1636C>T (p.Arg546Ter) is a nonsense variant in APC, a gene where loss of function is the established mechanism for familial adenomatous polyposis.1 This variant introduces a premature termination codon at position 546, upstream of the last exon-exon junction, predicting nonsense-mediated decay and complete loss of the C-terminal 2297 amino acids including all β-catenin binding and regulatory domains.2 The variant is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (1/1,613,730 alleles; AF=6.2×10⁻⁷), meeting PM2_Supporting under APC VCEP v2.1.0.3 ClinVar classifies this variant as Pathogenic based on submissions from multiple clinical laboratories, though individual case-level phenotype annotation is not publicly available for PS4 assessment.4 SpliceAI predicts no cryptic splice impact (max delta=0.01), consistent with this being a straightforward null variant rather than a splicing defect.5
APC
Final classification
Likely Pathogenic
APC c.1636C>T · p.Arg546Ter
APC
NM_001127511.3:c.1636C>T (p.Arg546Ter) is a nonsense variant in APC, a gene where loss of function is the established mechanism for familial adenomatous polyposis.
ClinGen InSiGHT Hereditary Colorectal Cancer/Polyposis Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for APC Version 2.1.0 v2.1.0 criteria-combination framework: matched Rule20 (1 Pathogenic.Very Strong + 1 Pathogenic.Supporting) with applied criteria: PVS1 very strong, PM2 supporting; maps to Likely Pathogenic.
Classification rationale
PVS1PM2
Likely Pathogenic
APC c.1636C>T
PVS1 + PM2
→
Likely Pathogenic
Gene diagram
· NM_001127511.3 · variants mapped to exon structure
APC
NM_001127511.3
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 14 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PVS1
very strong
Pathogenic
NM_001127511.3:c.1636C>T is a nonsense variant (p.Arg546Ter) in APC, a gene where loss of function is the established disease mechanism for familial adenomatous polyposis. The premature termination codon at position 546 lies upstream of the last exon-exon junction (c.1904/1905) by 269 nucleotides, well beyond the 50-55 nucleotide NMD boundary, predicting nonsense-mediated decay. Under the ClinGen InSiGHT APC VCEP v2.1.0, null variants in APC qualify for PVS1.
Nonsense variant p.Arg546Ter creates a premature termination codon in exon 12 of 14APC is a gene where LOF is the predominant disease mechanism per APC VCEP v2.1.0Variant is upstream of the last exon-exon junction with NMD predicted (PMC6185798)
✓
PM2
supporting
Pathogenic
NM_001127511.3:c.1636C>T is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (1/1,613,730 alleles; AF=6.2e-7). Under the APC VCEP v2.1.0, PM2_Supporting is met when the allele count is ≤1 and the allele frequency is <0.001% (0.00001). The observed AF of 0.000062% is below this threshold.
Absent from gnomAD v2.1gnomAD v4.1: AC=1AF=6.2e-7 (0.000062%)
Assessed · not applied
Pathogenic
PS1
PS1 requires the same amino acid change as a previously established pathogenic variant.
PS2
No de novo occurrence with confirmed paternity and maternity has been identified for NM_001127511.3:c.1636C>T in the available literature, ClinVar submissions, or exploratory search results.
PS3
No well-established functional study directly evaluating NM_001127511.3:c.1636C>T was identified.
PS4
ClinVar lists NM_001127511.3:c.1636C>T as Pathogenic with clinical laboratory submissions, but disaggregated case counts with phenotype points per the APC VCEP Table 1 are not available from the current evidence.
PM6
No assumed de novo observation (without confirmation of paternity and maternity) has been identified for NM_001127511.3:c.1636C>T in the available literature, ClinVar submissions, or exploratory search results.
PP1
No co-segregation data has been identified for NM_001127511.3:c.1636C>T in the available literature or databases.
PP3
Under the APC VCEP v2.1.0, PP3 is applicable only to missense variants (via in silico splicing predictors to reveal possible splice effects) and non-canonical splicing variants.
Benign
BA1
Under the APC VCEP v2.1.0, BA1 requires a gnomAD Popmax Filtering AF ≥0.1% (0.001).
BS1
Under the APC VCEP v2.1.0, BS1 requires a gnomAD Popmax Filtering AF ≥0.001% (0.00001).
BS2
No observation of NM_001127511.3:c.1636C>T in healthy individuals aged ≥50 years with documented absence of polyposis has been identified.
BS3
BS3 under the APC VCEP requires RNA assay evidence showing no mRNA aberration or protein assay showing retention of wild-type function.
BS4
No evidence for lack of segregation of NM_001127511.3:c.1636C>T with FAP in affected family members has been identified.
BP2
No observation of NM_001127511.3:c.1636C>T in trans with a (likely) pathogenic APC variant has been reported.
BP5
No alternate genetic basis for the colorectal polyposis phenotype (e.g., pathogenic variants in MUTYH, NTHL1, MSH3, POLD1, POLE) has been documented in individuals carrying NM_001127511.3:c.1636C>T.
N/A · 10
PM1 · PM5 · PP2 · PP4 · PP5 · BP1 · BP3 · BP4 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 6.19682e-07; MAF= 0.00006%, 1/1613730 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.47596e-07; MAF= 0.00008%, 1/1179808 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,613,730
0 hom
0 hom
European (non-Finnish) 1 / 1,179,808 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). BayesDel score = 0.66.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Loss-of-function; curated oncogenicity label: Likely Oncogenic.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 9 PMIDs not cited in assessment
11062151 ↗
Expression of beta-catenin and full-length APC protein in normal and neoplastic colonic tissues.
ONCOKB
15561772 ↗
Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability.
ONCOKB
1324223 ↗
Eight novel inactivating germ line mutations at the APC gene identified by denaturing gradient gel electrophoresis.
CLINVAR
14523376 ↗
Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis.
CLINVAR
15833136 ↗
Rare mutations predisposing to familial adenomatous polyposis in Greek FAP patients.
CLINVAR
23159591 ↗
APC germline mutations in individuals being evaluated for familial adenomatous polyposis: a review of the Mayo Clinic experience with 1591 consecutive tests.
CLINVAR
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR