NM_001128849.1:c.3484G>A (p.Gly1162Ser) is a missense variant in the SMARCA4 ATPase/helicase C-terminal domain, a well-established critical functional domain where pathogenic missense variants are enriched (PM1).1 The variant is absent from all population databases, including gnomAD v2.1, v4.1, and gnomAD-Canada, with allele frequency of 0 (PM2).2 A different missense change at the same codon, p.Gly1162Cys, has been experimentally demonstrated to cause loss of function in a systematic SMARCA4 functional characterization study (PM5).3 Multiple lines of in silico evidence support a deleterious effect: REVEL score of 0.969 and BayesDel score of 0.592 both predict damaging consequences (PP3).4 Per generic ACMG/AMP 2015 classification rules (PMID:25741868), the combination of three moderate criteria (PM1 + PM2 + PM5) meets the threshold for Likely Pathogenic.5
SMARCA4
Final classification
Likely Pathogenic
SMARCA4 c.3484G>A · p.Gly1162Ser
SMARCA4
NM_001128849.1:c.3484G>A (p.Gly1162Ser) is a missense variant in the SMARCA4 ATPase/helicase C-terminal domain, a well-established critical functional domain where pathogenic missense variants are enriched (PM1).
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 moderate, PM2 moderate, PM5 moderate, PP3 supporting; combination = 3 moderate + 1 supporting, which maps to Likely Pathogenic.
Classification rationale
PM1PM2PM5PP3
Likely Pathogenic
SMARCA4 c.3484G>A
PM1 + PM2 + PM5 + PP3
→
Likely Pathogenic
4
revelbayesdel
5
generic_acmg_combination_rules
Gene diagram
· NM_001128849.1 · variants mapped to exon structure
SMARCA4
NM_001128849.1
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 21 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
p.Gly1162Ser is located within the SMARCA4 ATPase/helicase C-terminal domain, a well-established critical functional domain. PMID:33144586 characterized multiple missense mutations across this domain (including adjacent residues G1159V and G1162C) and demonstrated that mutations in this region abrogate chromatin remodeling activity and are loss-of-function. The helicase domain is essential for SMARCA4 catalytic activity and is a known hotspot for pathogenic missense variants in Coffin-Siris syndrome and cancer predisposition.
Variant falls within the SMARCA4 ATPase/helicase C-terminal domainfunctionally characterized as critical by PMID:33144586. Multiple pathogenic missense mutations cluster in this region.
✓
PM2
moderate
Pathogenic
NM_001128849.1:c.3484G>A is absent from all population databases: gnomAD v2.1 (AF=0), gnomAD v4.1 (AF=0), and gnomAD-Canada v1.0 (AF=0). This meets PM2 (<0.1% allele frequency threshold per generic ACMG).
Absent from gnomAD v2.1 (exomes)gnomAD v4.1 (exomes/genomes)and gnomAD-Canada v1.0 (AF=0.0).
✓
PM5
moderate
Pathogenic
A different missense change at the same codon, p.Gly1162Cys (c.3484G>T), was directly tested in PMID:33144586 and demonstrated to be loss-of-function. G1162C-expressing cells failed to rescue SMARCA2 depletion, had no detectable nucleosome remodeling activity, and exhibited dominant-negative chromatin effects. No ClinVar PM5 comparators were identified at this residue, but the literature provides functional evidence that a different missense at Gly1162 is pathogenic, satisfying PM5.
p.Gly1162Cys at the same codon was functionally tested and demonstrated LOF (PMID:33144586).
✓
PP3
supporting
Pathogenic
Multiple lines of in silico evidence support a deleterious effect: REVEL score 0.969 (strongly pathogenic), BayesDel score 0.592 (elevated). SpliceAI predicts no splice impact (max delta score 0.00), consistent with a missense effect rather than splicing alteration. Two independent computational methods agree on a damaging prediction.
REVEL=0.969 (high-confidence damaging prediction). BayesDel=0.592 (elevated). SpliceAI max delta=0.00 (no splice effect).
Assessed · not applied
Pathogenic
PVS1
PVS1 is not applicable: NM_001128849.1:c.3484G>A is a missense variant (p.Gly1162Ser).
PS1
No alternative nucleotide change producing the same p.Gly1162Ser amino acid substitution has been reported as pathogenic in ClinVar or the literature.
PS2
No de novo occurrence data is available for this variant.
PS3
The exact variant p.Gly1162Ser was not directly tested in any functional study.
PS4
No case-control data comparing variant prevalence in affected versus unaffected individuals is available.
PM6
No de novo occurrence data with confirmed paternity and maternity is available for this variant.
PP1
No co-segregation data is available for this variant.
PP2
SMARCA4 has a mixed mutational spectrum: truncating variants cause rhabdoid tumor predisposition syndrome type 2 (RTPS2), while missense variants in the helicase domain cause Coffin-Siris syndrome (CSS).
PP4
No patient phenotype or family history data is available for assessment.
PP5
ClinVar review status is 'criteria provided, single submitter' (1-star).
Benign
BA1
The variant is absent from all population databases (gnomAD v2.1, v4.1, Canada).
BS1
The variant is absent from all population databases.
BS2
No observation in healthy adult individuals has been reported for this variant.
BS3
No well-established functional studies demonstrate a benign effect.
BS4
No segregation data in affected families is available to evaluate lack of segregation.
BP1
BP1 does not apply: SMARCA4 disease is caused by both missense and truncating variants.
BP2
No observation in trans with a pathogenic variant has been reported.
BP4
Computational predictions are unanimously deleterious: REVEL 0.969 (strongly pathogenic), BayesDel 0.592 (elevated).
BP5
No alternate molecular basis for disease has been identified in a case harboring this variant.
BP6
ClinVar classification is 'Tier I - Strong' (pathogenic/likely pathogenic), not benign.
BP7
BP7 does not apply: c.3484G>A is a missense variant (p.Gly1162Ser), not a synonymous/silent variant.
Research & evidence
Population frequency · supports pathogenic
gnomAD v4.1
gnomAD v2.1
v4.1
Absent from gnomAD v4.1.
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
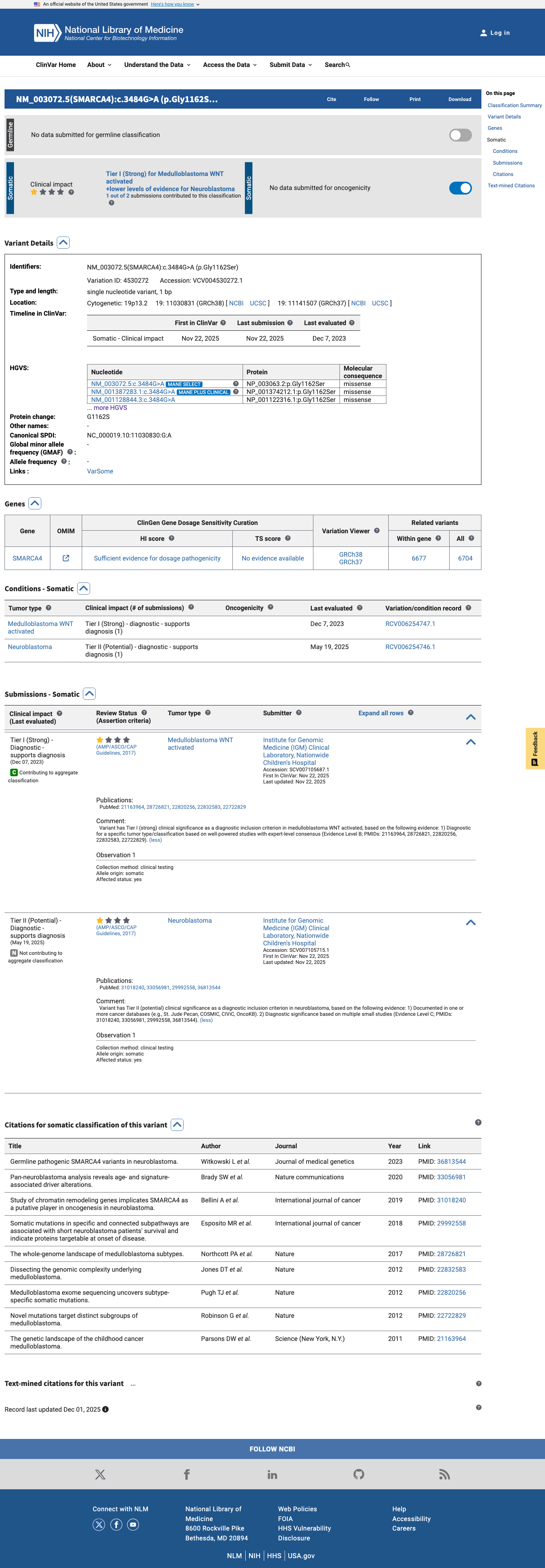
ClinVar
This variant has been reported in ClinVar but submission details could not be extracted. (ClinVarID = 4530272)
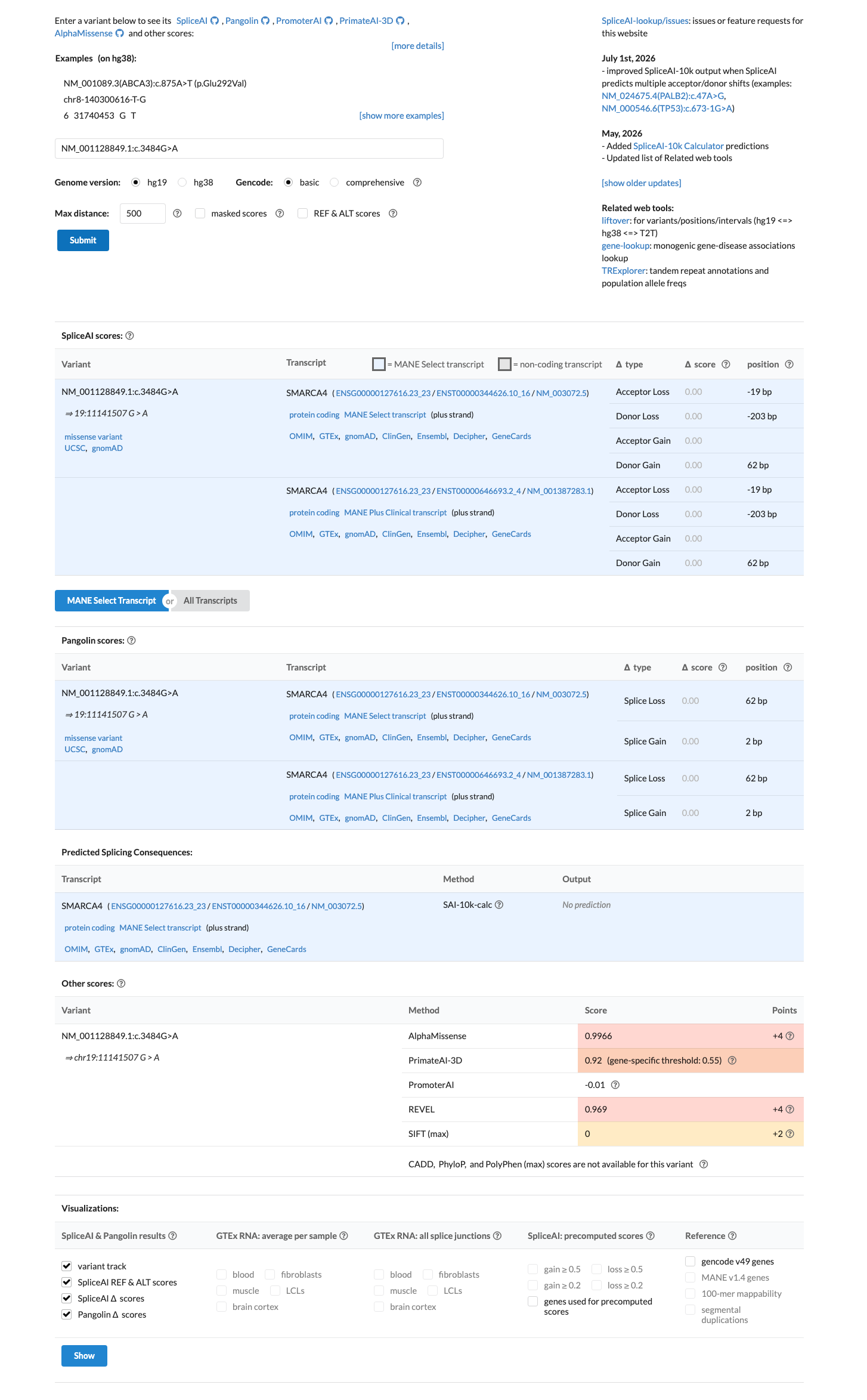
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.969. BayesDel score = 0.591961.
Functional
Likely Oncogenic
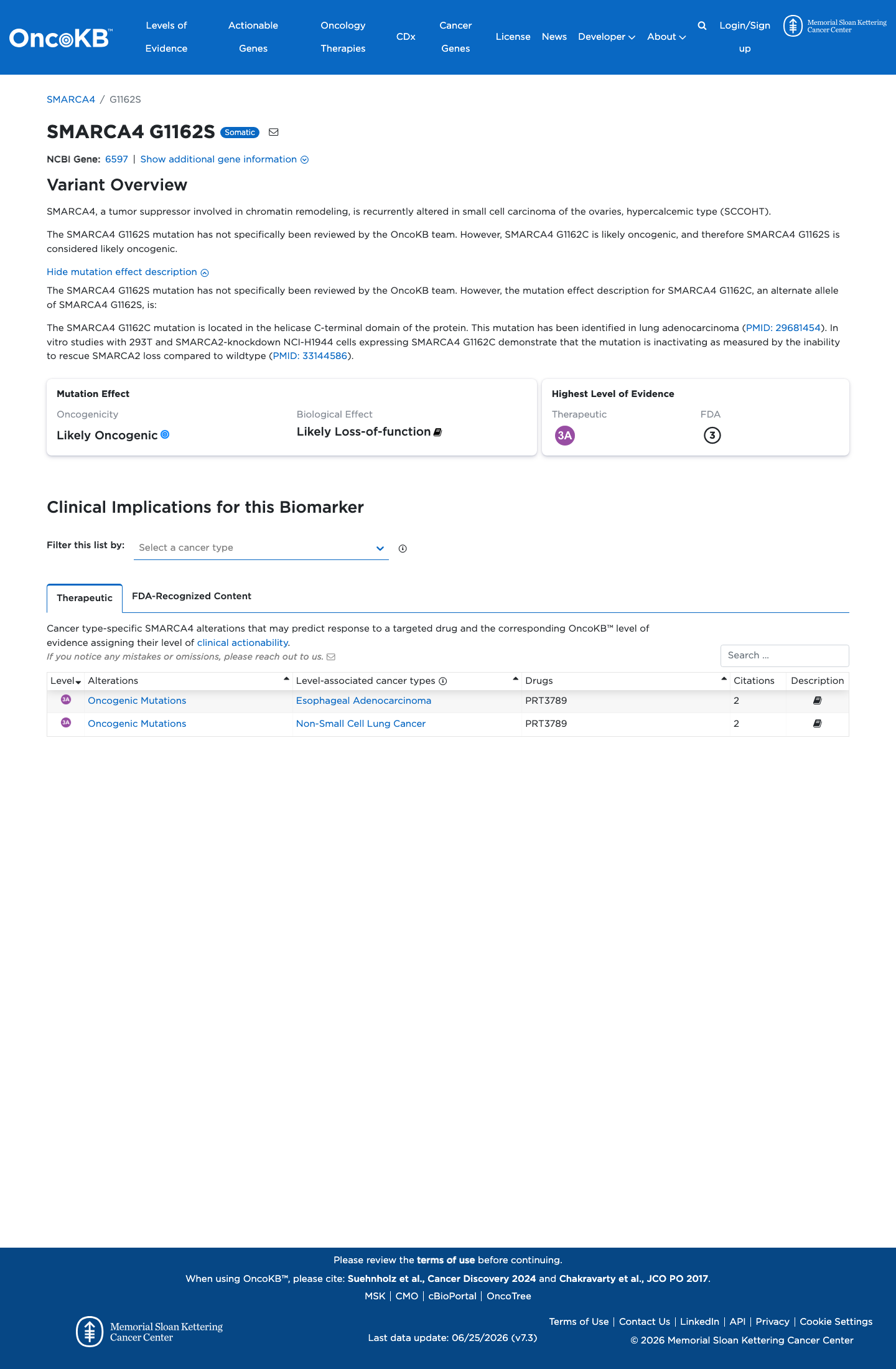
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. SMARCA4, a tumor suppressor involved in chromatin remodeling, is recurrently altered in small cell carcinoma of the ovaries, hypercalcemic type (SCCOH
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
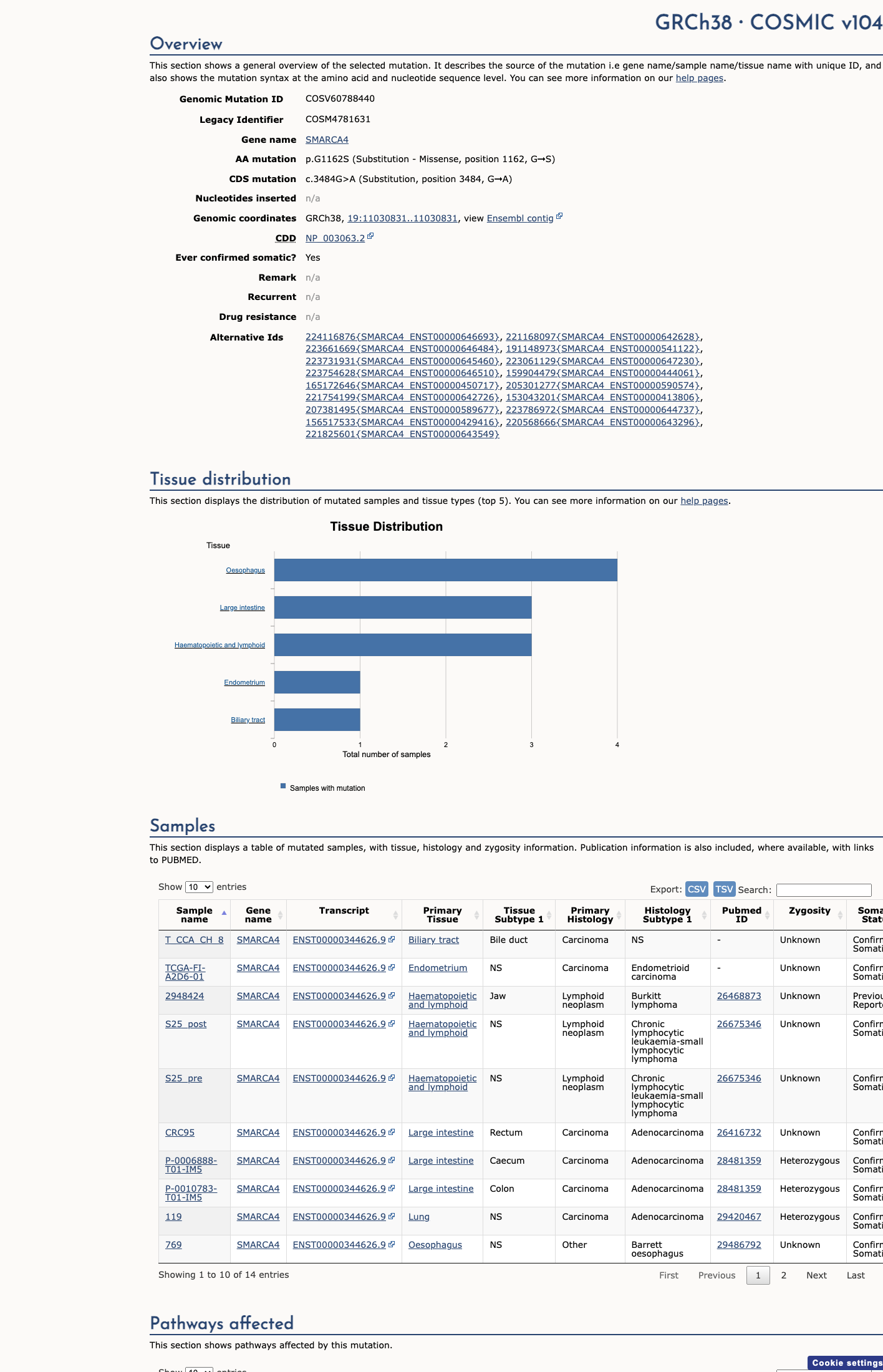
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV60788440, n = 14 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 6 further PMIDs triaged but not cited — see Sources & References.
Functional characterization of SMARCA4 variants identified by targeted exome-sequencing of 131,668 cancer patients.
Searched
c.3484G>Ap.Gly1162SerG1162SG1162C
Found
Systematic functional characterization of SMARCA4 missense variants from 131,668 cancer patients. The study tested 14 helicase domain variants including G1162C at the same codon (Gly1162), which demonstrated complete loss of chromatin remodeling activity, inability to induce gene expression, and failure to rescue SMARCA2 depletion. G1162S was not directly tested or reported. The study provides domain-level evidence for PM1 and same-codon evidence for PM5.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
→PM5 supports · met
Why
G1162C (same codon, different amino acid) confirmed LOF; supports PM5 (same codon) and PM1 (helicase domain critical). G1162S was not directly tested.
The majority of SMARCA4 mutants tested were unable to rescue SMARCA2 knockdown, confirming that these mutants (K785R, E882K, T910M, R1135W, G1162C, R1192C, G1232S) are indeed loss-of-function (LOF).
Location Results: 'SMARCA4 missense mutants have reduced remodeling activity'; Fig. 5a; cell line NCI-H1435 harbors homozygous G1162C · Context FRET nucleosome remodeling assay, ATAC-seq, qChIP, and clonogenic growth rescue in SMARCA4-deficient NCI-H1944 and A549 cell lines · full text
Sources & reference links
Triaged references · 6 PMIDs not cited in assessment
31018240 ↗
Study of chromatin remodeling genes implicates SMARCA4 as a putative player in oncogenesis in neuroblastoma.
CLINVAR