NM_001145661.1:c.710G>A (p.Gly237Asp) in GATA2 is classified as Benign based on generic ACMG/AMP 2015 criteria. This variant is present in gnomAD v2.1 at an allele frequency of 0.366% (1033/282138 alleles, including 17 homozygotes), exceeding the BS1 threshold of 0.3% for a rare dominant disorder.1 Seventeen homozygous individuals are observed in gnomAD v2.1 (37 in gnomAD v4.1). Homozygosity for a variant in a gene causing a highly penetrant dominant disorder (GATA2 deficiency) is strong evidence of a benign effect (BS2).2 Multiple in silico predictors support a benign interpretation: SpliceAI predicts no splice impact (max delta 0.00), REVEL score is 0.401 (below 0.5 threshold), and BayesDel score is -0.00217913 (benign range) (BP4).3 Eight clinical laboratories in ClinVar classify this variant as Likely benign (4) or Benign (4), consistent with a benign interpretation (BP6).4
GATA2
Final classification
Benign
GATA2 c.710G>A · p.Gly237Asp
GATA2
NM_001145661.1:c.710G>A (p.Gly237Asp) in GATA2 is classified as Benign based on generic ACMG/AMP 2015 criteria.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: BS1 strong benign, BS2 strong benign, BP4 supporting benign, BP6 supporting benign; combination = 2 strong benign + 2 supporting benign, which maps to Benign.
Classification rationale
BS1BS2BP4BP6
Benign
GATA2 c.710G>A
BS1 + BS2 + BP4 + BP6
→
Benign
Gene diagram
· NM_001145661.1 · variants mapped to exon structure
GATA2
NM_001145661.1
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 18 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
BS1
strong
Benign
The variant has a total allele frequency of 0.366% (1033/282138 alleles) in gnomAD v2.1, which exceeds the 0.3% BS1 threshold. This frequency is greater than expected for GATA2 deficiency, a rare dominant disorder.
gnomAD v2.1 total AF 0.366% (> 0.3% BS1 threshold)1033 alleles17 homozygotes
✓
BS2
strong
Benign
Seventeen homozygotes are observed in gnomAD v2.1 (37 in gnomAD v4.1). GATA2 deficiency is a dominant disorder with high penetrance; the presence of homozygous individuals in a population database is incompatible with a pathogenic variant in this gene.
gnomAD v2.1: 17 homozygotesgnomAD v4.1: 37 homozygotes. Homozygosity for a dominant disease gene is strong evidence of benign impact.
✓
BP4
supporting
Benign
Multiple computational predictors suggest no impact: SpliceAI max delta 0.00 (no predicted splice alteration), REVEL score 0.401 (below 0.5 threshold for predicted damaging), and BayesDel score -0.00217913 (in the benign range).
SpliceAI max delta 0.00REVEL 0.401BayesDel -0.00217913 (benign). All three in silico predictors are consistent with a benign variant.
✓
BP6
supporting
Benign
Eight clinical laboratory submissions in ClinVar (ID 343139) classify this variant as Likely benign (4 laboratories) or Benign (4 laboratories). Although review status is 'criteria provided, single submitter' per submission, the consensus across multiple independent clinical laboratories supports a benign interpretation.
ClinVar ID 343139: 4 laboratories report Likely benign4 report Benign. Submitters include InvitaeAmbry Genetics
Assessed · not applied
Pathogenic
PS1
No evidence of a different nucleotide change at the same amino acid residue (Gly237) resulting in a known pathogenic variant.
PS2
No de novo data available for this variant.
PS3
No variant-specific functional studies were identified.
PS4
The variant is present at 0.366% total allele frequency in gnomAD v2.1, which is inconsistent with a rare pathogenic variant.
PM1
The variant does not lie in a statistically significant mutational hotspot per cancerhotspots.org.
PM2
Variant is present in gnomAD v2.1 at total allele frequency 0.366% (1033/282138 alleles), exceeding the 0.1% PM2 threshold for rarity.
PM6
No de novo data available for this variant.
PP1
No co-segregation data available for this variant.
PP2
Although GATA2 missense variants are a known disease mechanism in GATA2 deficiency syndrome, this specific variant is present at high population frequency (3.76% in Finnish, 17 homozygotes), which is incompatible with a pathogenic missense variant in a dominant disorder gene.
PP3
Multiple in silico predictors suggest no damaging effect: REVEL score 0.401 (below 0.5 threshold), BayesDel score -0.00217913 (benign range), SpliceAI max delta 0.00 (no predicted splice impact).
PP4
No patient phenotype information available to assess phenotypic specificity.
PP5
ClinVar reports this variant as Likely benign (4 clinical laboratories) and Benign (4 clinical laboratories).
Benign
BA1
Total allele frequency in gnomAD v2.1 is 0.366%, below the 1% BA1 threshold.
BS3
No variant-specific well-established functional studies demonstrating no damaging effect were identified.
BS4
No segregation data available to assess lack of co-segregation with disease.
BP1
GATA2 deficiency is caused by both missense and truncating variants.
BP2
No data on whether this variant has been observed in trans with a known pathogenic GATA2 variant.
BP5
No information on whether this variant has been observed in cases with an alternate molecular basis for disease.
N/A · 6
PVS1 · PM3 · PM4 · PM5 · BP3 · BP7
Research & evidence
Population frequency · supports benign
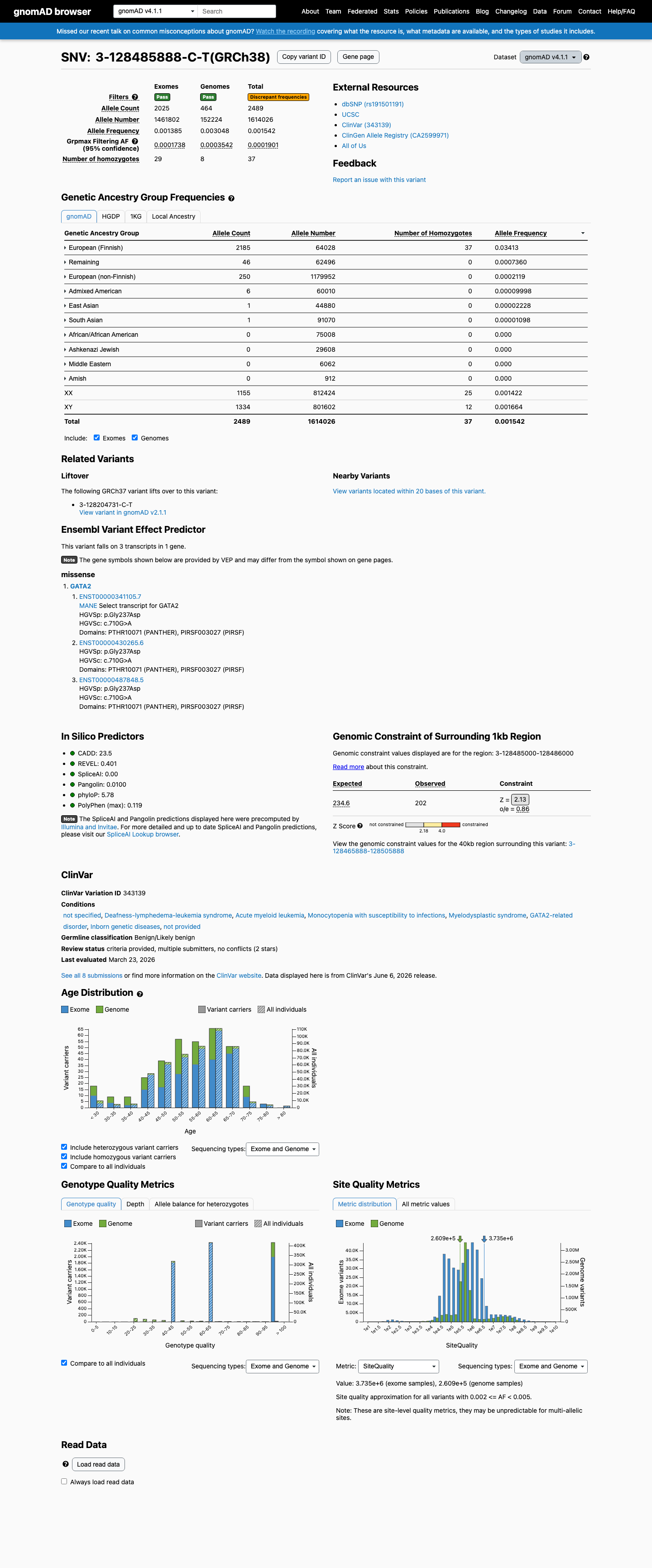
gnomAD v4.1
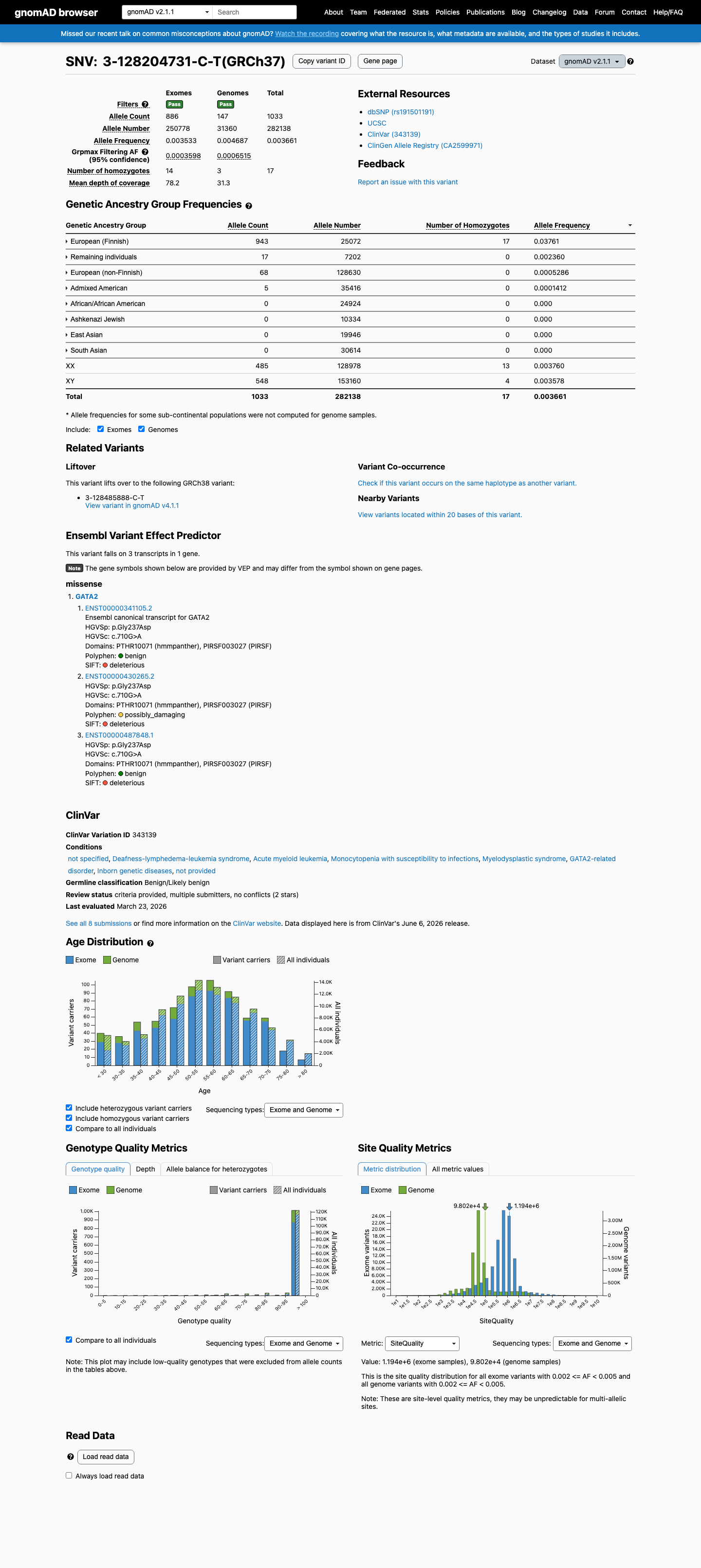
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.00154211; MAF= 0.15421%, 2489/1614026 alleles, homozygotes = 37) and has highest observed frequency in the European (Finnish) population (AF= 0.0341257; MAF= 3.41257%, 2185/64028 alleles, homozygotes = 37); grpmax FAF= 0.00019008.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.00366133; MAF= 0.36613%, 1033/282138 alleles, homozygotes = 17) and has highest observed frequency in the European (Finnish) population (AF= 0.0376117; MAF= 3.76117%, 943/25072 alleles, homozygotes = 17); grpmax FAF= 0.00065149.
🇨🇦 CA
This variant is present in gnomAD-Canada v1.0 (AF= 0.00021734405564007825, 4/18404 alleles, homozygotes = 0).
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.15%
· 2489 / 1,614,026
37 hom · FAF 0.019%
37 hom · FAF 0.019%
European (Finnish) 2185 / 64,028 |
3.4% 37 hom |
Remaining individuals 46 / 62,496 |
0.074% |
European (non-Finnish) 250 / 1,179,952 |
0.021% |
Admixed American 6 / 60,010 |
0.01% |
East Asian 1 / 44,880 |
0.0022% |
South Asian 1 / 91,070 |
0.0011% |
+ 4 not observed (Amish, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.37%
· 1033 / 282,138
17 hom · FAF 0.065%
17 hom · FAF 0.065%
European (Finnish) 943 / 25,072 |
3.8% 17 hom |
Remaining individuals 17 / 7,202 |
0.24% |
European (non-Finnish) 68 / 128,630 |
0.053% |
Admixed American 5 / 35,416 |
0.014% |
+ 4 not observed (African/African American, Ashkenazi Jewish, East Asian, South Asian)
gnomAD Canada 🇨🇦
0.022%
· 4 / 18,404
0 hom · FAF 0.003%
0 hom · FAF 0.003%
Remaining individuals 2 / 1,136 |
0.18% |
European (non-Finnish) 2 / 11,732 |
0.017% |
+ 7 not observed (African/African American, Latino/Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Middle Eastern, South Asian)
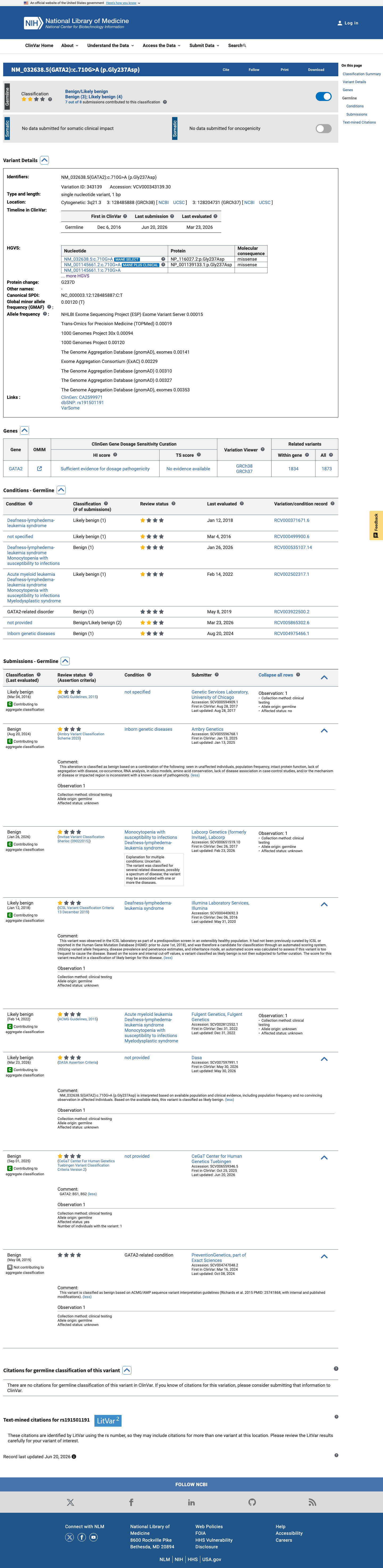
ClinVar
This variant has been reported in ClinVar as Likely benign (4 clinical laboratories) and as Benign (4 clinical laboratories). (ClinVarID = 343139)
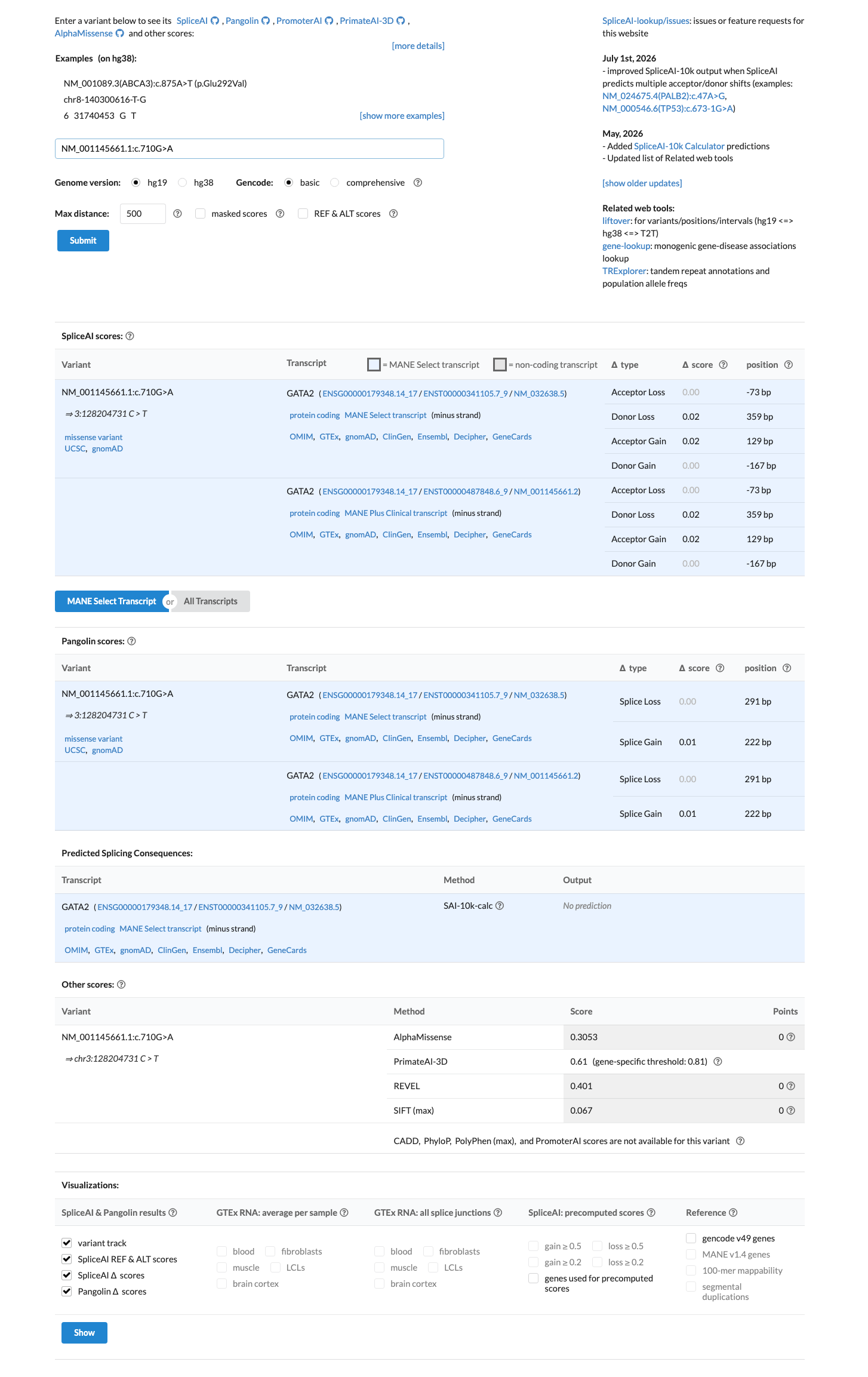
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.401. BayesDel score = -0.00217913.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. GATA2, a transcription factor, is recurrently mutated in hematological malignancies and various solid tumors.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
32171751 ↗
Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up.
CLINVAR
23037933 ↗
Including the initial newborn screening bloodspot collection device serial number on birth certificates: basis and recommendations from the Secretary of Health and Human Services' Advisory Committee on Heritable Disorders in Newborns and Children.
CLINVAR
24394680 ↗
Parental permission for pilot newborn screening research: guidelines from the NBSTRN.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR