NM_001277115.2:c.4879C>T (p.Arg1627Cys) is a missense variant in DNAH11, a gene associated with autosomal recessive primary ciliary dyskinesia (PCD) and heterotaxy syndromes. This variant is extremely rare in population databases: gnomAD v2.1 AF = 0.00107% (3/280,426 alleles), gnomAD v4.1 AF = 0.00155% (25/1,613,592 alleles), and it is absent from gnomAD-Canada, with no homozygotes observed in any population database (PM2_supporting).1 In silico prediction tools support a deleterious effect, with a REVEL score of 0.887 exceeding the damaging threshold (PP3_supporting).2 SpliceAI predicts no splicing impact (max delta score = 0.00), and the variant does not lie in a statistically significant mutational hotspot.3 This variant has been reported in ClinVar with conflicting classifications: four clinical laboratories classify it as Uncertain Significance, one as Pathogenic, and one as Likely Pathogenic. No expert panel review has been performed (ClinVar Variation ID: 163104).4 The variant has been observed somatically in COSMIC (COSV60977423, n = 2). No variant-specific functional data, case-control studies, segregation data, or de novo reports were identified in the reviewed literature. The five publications cited by ClinVar submitters are either methodology/guideline papers (PMID:24033266, PMID:25741868, PMID:28492532), a general GeneReviews overview (PMID:20301301), or a PCD cohort study that does not specifically mention c.4879C>T (PMID:31772028).5
DNAH11
Final classification
VUS
DNAH11 c.4879C>T · p.Arg1627Cys
DNAH11
NM_001277115.2:c.4879C>T (p.Arg1627Cys) is a missense variant in DNAH11, a gene associated with autosomal recessive primary ciliary dyskinesia (PCD) and heterotaxy syndromes.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM2 supporting, PP3 supporting; combination = 2 supporting, which maps to VUS.
Classification rationale
PM2PP3
VUS
DNAH11 c.4879C>T
PM2 + PP3
→
VUS
Gene diagram
· NM_001277115.2 · variants mapped to exon structure
DNAH11
NM_001277115.2
Fetching transcript structure from UCSC…
Applied criteria · 2 applied · 21 assessed
Applied · 2
Strength
Supporting
Moderate
Strong
Very strong
✓
PM2
supporting
Pathogenic
This variant is absent or extremely rare in population databases. gnomAD v2.1 AF = 0.00107% (3/280,426 alleles), gnomAD v4.1 AF = 0.00155% (25/1,613,592 alleles; grpmax FAF = 1.221e-05), and it is absent from gnomAD-Canada. All population frequencies are well below the 0.1% PM2 threshold. No homozygotes have been observed.
gnomAD v2.1 AF = 1.0698e-05 (3/280426 alleles0 homozygotes)
✓
PP3
supporting
Pathogenic
In silico prediction tools support a deleterious effect: REVEL score is 0.887, which exceeds the 0.75 threshold for damaging prediction. BayesDel score is 0.43 (intermediate). SpliceAI predicts no splicing impact (max delta = 0.00). The REVEL score provides supporting computational evidence of pathogenicity.
REVEL score = 0.887 (>0.75 damaging threshold)BayesDel score = 0.430511SpliceAI max delta = 0.00 (no splice impact)
Assessed · not applied
Pathogenic
PS1
No evidence of an alternative nucleotide change at codon 1627 that results in the same p.Arg1627Cys missense and has been established as pathogenic.
PS2
No de novo occurrence data with confirmed maternity/paternity is available for this variant.
PS3
No experimental functional data was identified for NM_001277115.2:c.4879C>T (p.Arg1627Cys) in any reviewed publication.
PS4
No variant-specific case-control or cohort data was identified.
PM1
The variant does not lie within a statistically significant mutational hotspot as assessed by cancerhotspots.org.
PM5
Automated PM5 candidate harvesting did not identify any same-residue comparator variants at position 1627 in DNAH11 with a different amino acid change classified as pathogenic.
PM6
No de novo occurrence has been reported for this variant in any ClinVar submission or reviewed publication.
PP1
No co-segregation data is available for this variant.
PP2
PP2 requires a gene with a low rate of benign missense variation where missense variants are a common disease mechanism.
PP4
No patient-specific phenotypic data is available for this variant.
PP5
Although one clinical laboratory (Fulgent Genetics) classified this variant as Likely Pathogenic and another as Pathogenic, four additional clinical laboratories classified it as Uncertain Significance.
Benign
BA1
The maximum population allele frequency is 0.00155% in gnomAD v4.1, which is well below the 1% BA1 threshold.
BS1
The maximum population allele frequency is 0.00333% in the Admixed American subpopulation (gnomAD v4.1), which is well below the 0.3% BS1 threshold.
BS2
No evidence that this variant has been observed in a healthy adult individual in a context sufficient to meet BS2.
BS3
No functional studies demonstrating a benign or normal effect for this variant were identified in the literature.
BS4
No segregation data is available to evaluate lack of co-segregation with disease.
BP1
BP1 applies when a missense variant occurs in a gene where only truncating variants cause disease.
BP2
No evidence that this variant has been observed in trans with a known pathogenic variant in DNAH11 in an unaffected individual.
BP4
Multiple in silico prediction tools support a deleterious effect: REVEL score is 0.887 (damaging), BayesDel score is 0.43 (intermediate, not clearly benign).
BP5
No evidence that this variant has been observed in a case with an alternate molecular basis for disease.
BP6
No reputable source reports this variant as benign.
N/A · 2
PVS1 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
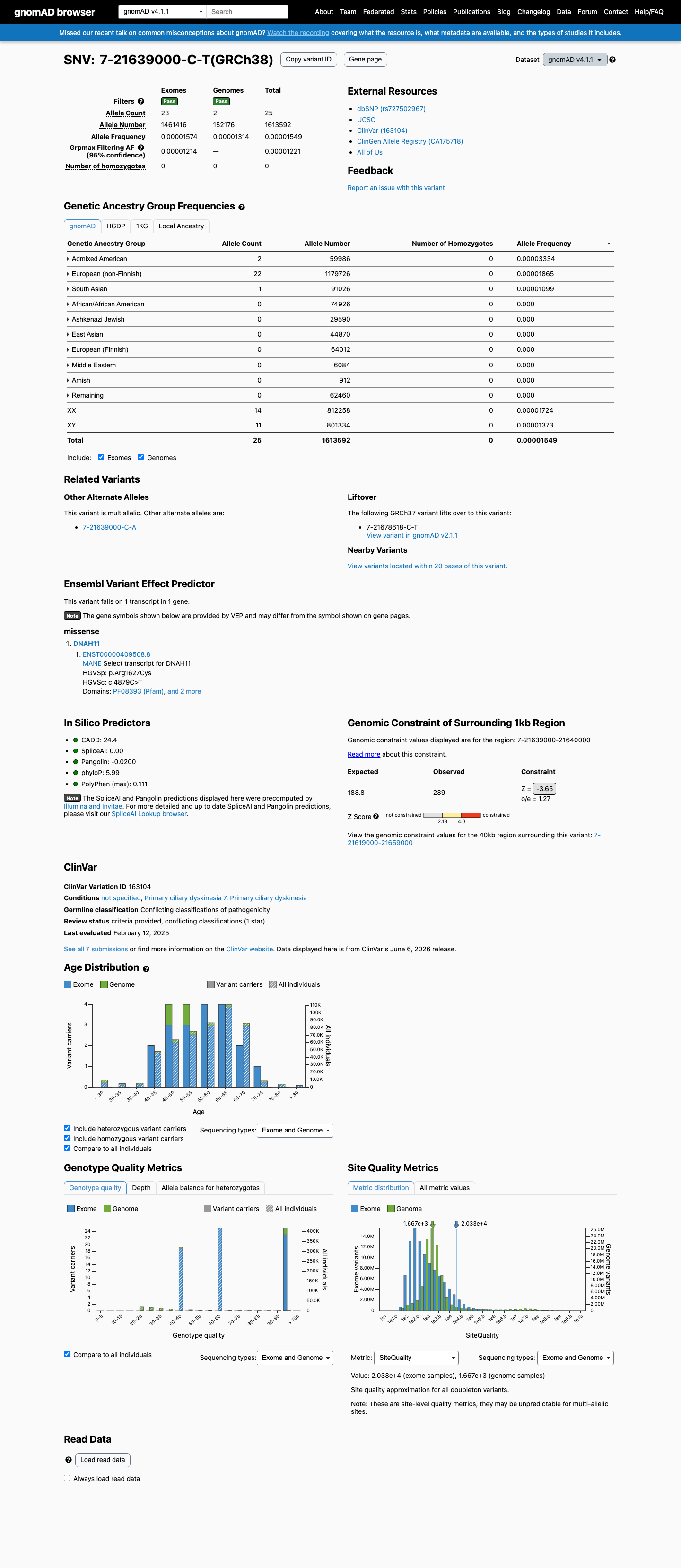
This variant is present in gnomAD v4.1 (AF= 1.54934e-05; MAF= 0.00155%, 25/1613592 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 3.33411e-05; MAF= 0.00333%, 2/59986 alleles, homozygotes = 0); grpmax FAF= 1.221e-05.
v2.1
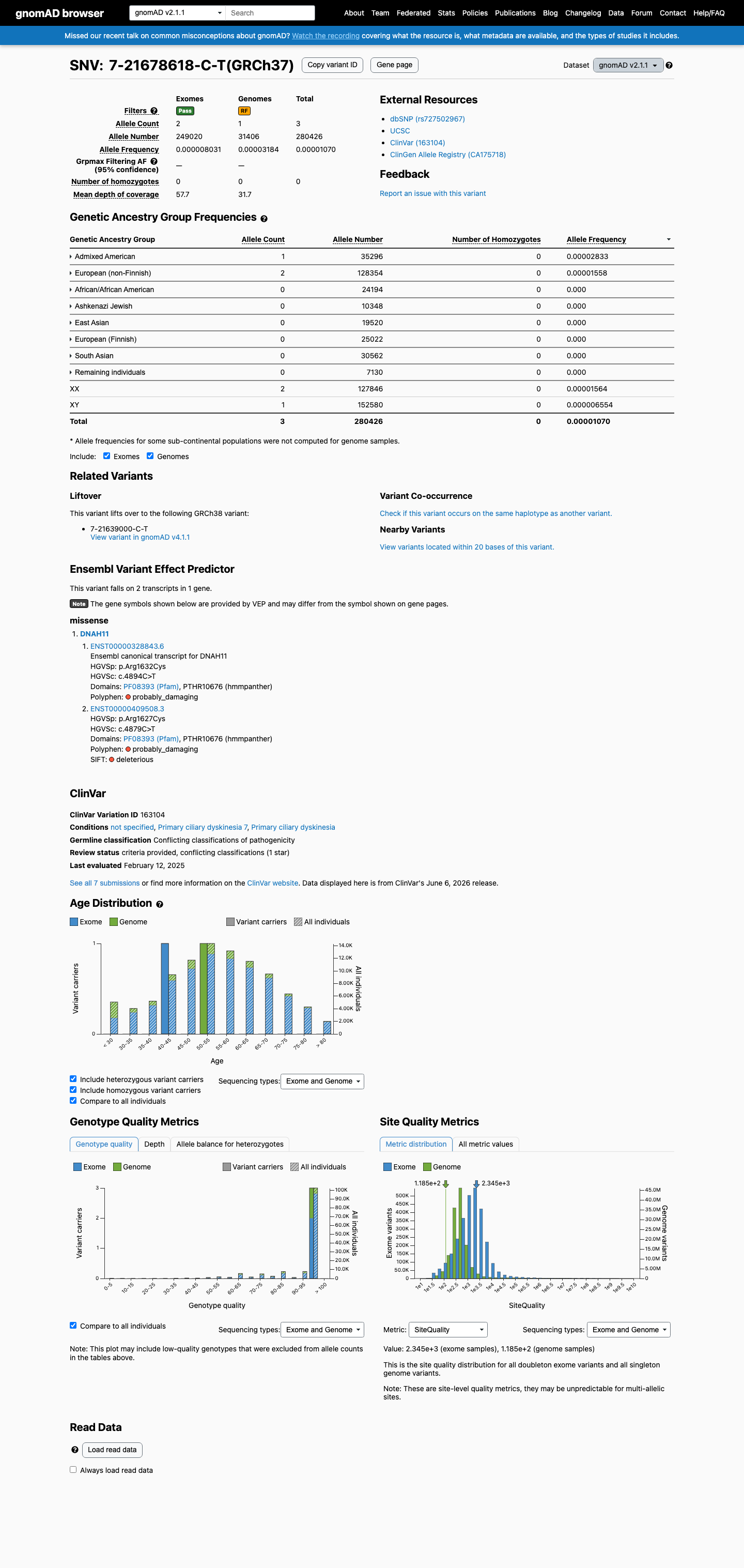
This variant is present in gnomAD v2.1 (AF= 1.0698e-05; MAF= 0.00107%, 3/280426 alleles, homozygotes = 0) and has highest observed frequency in the Admixed American population (AF= 2.83318e-05; MAF= 0.00283%, 1/35296 alleles, homozygotes = 0).
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0015%
· 25 / 1,613,592
0 hom · FAF 0.0012%
0 hom · FAF 0.0012%
Admixed American 2 / 59,986 |
0.0033% |
European (non-Finnish) 22 / 1,179,726 |
0.0019% |
South Asian 1 / 91,026 |
0.0011% |
+ 7 not observed (Remaining individuals, European (Finnish), Amish, East Asian, Middle Eastern, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0011%
· 3 / 280,426
0 hom
0 hom
Admixed American 1 / 35,296 |
0.0028% |
European (non-Finnish) 2 / 128,354 |
0.0016% |
+ 6 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
ClinVar
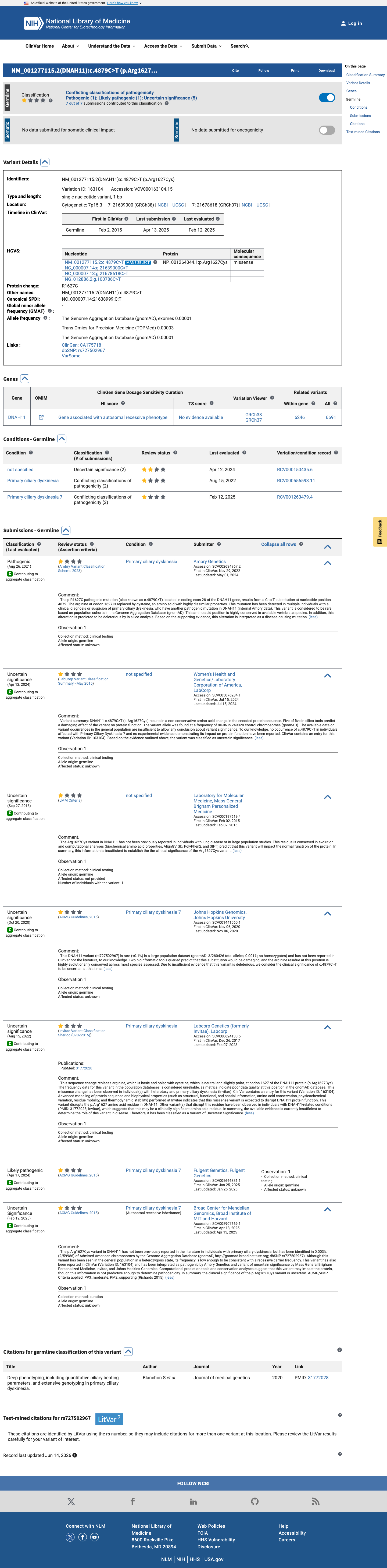
This variant has been reported in ClinVar as Uncertain significance (4 clinical laboratories) and as Pathogenic (1 clinical laboratory) and as Likely pathogenic (1 clinical laboratory). (ClinVarID = 163104)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.887. BayesDel score = 0.430511.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.

COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV60977423, n = 2 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
31772028 ↗
Deep phenotyping, including quantitative ciliary beating parameters, and extensive genotyping in primary ciliary dyskinesia.
CLINVAR