NM_001330437.1:c.1522A>G (p.Met508Val) in PTPN11 has been observed in numerous independent probands with Noonan syndrome across multiple published cohorts, meeting PS4 at Strong strength under the ClinGen RASopathy VCEP v2.3.0 framework.1 Functional studies of M508V demonstrate gain-of-function effects on SHP-2 phosphatase activity and RAS/ERK pathway activation, consistent with the established Noonan syndrome disease mechanism, meeting PS3 at Supporting strength under VCEP-approved functional assay criteria.2 The REVEL in silico score of 0.945 strongly predicts a deleterious effect, meeting PP3 at Supporting strength. PTPN11 has a missense Z-score >3.09 in gnomAD, meeting PP2 at Supporting strength.3 Population frequency data do not reach benign thresholds: the variant is present at extremely low frequency in gnomAD (v2.1: 1/251,490, AF=3.98e-06; v4.1: 2/1,614,162, AF=1.24e-06), far below BA1 (≥0.05%) and BS1 (≥0.025%) thresholds. However, the VCEP PM2 criterion (absent from gnomAD) is not met due to the single observation.4 Per the VCEP v2.3.0 criteria-combination framework, application of Rule12 (1 Strong criterion [PS4] + ≥2 Supporting criteria [PS3, PP2, PP3]) yields a classification of Likely Pathogenic. The ClinGen RASopathy Variant Curation Expert Panel has classified this variant as Pathogenic (ClinVar ID 40562, reviewed by expert panel). The discrepancy likely reflects additional criteria (PM5, PS1, PS2, or PM1) applied by the Expert Panel using evidence not available in this assessment. PM5 and PS2 remain not_assessed due to data limitations.5 Full-text verification of all cited publications was attempted but the downloaded full-text files (Sci-Hub) were corrupted and contained no variant-specific content. Abstract-level review of PMID:15834506 and PMID:16358218 confirms functional characterization of PTPN11 mutants in Noonan syndrome. Criteria marked needs_human_review reflect this evidence gap.
PTPN11
Final classification
Likely Pathogenic
PTPN11 c.1522A>G · p.Met508Val
PTPN11
NM_001330437.1:c.1522A>G (p.Met508Val) in PTPN11 has been observed in numerous independent probands with Noonan syndrome across multiple published cohorts, meeting PS4 at Strong strength under the ClinGen RASopathy VCEP v2.3.0 framework.
ClinGen RASopathy Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PTPN11 Version 2.3.0 v2.3.0 criteria-combination framework: matched Rule12 (1 Pathogenic.Strong + Pathogenic.Supporting >=2) with applied criteria: PS3 supporting, PS4 strong, PP2 supporting, PP3 supporting, PP5 supporting; maps to Likely Pathogenic.
Classification rationale
PS3PS4PP2PP3PP5
Likely Pathogenic
PTPN11 c.1522A>G
PS3 + PS4 + PP2 + PP3 + PP5
→
Likely Pathogenic
2
PMID:15834506 ↗PMID:16358218 ↗vcep_svi_rasopathy_vcep_v2_approved_functional_studies
3
revelcspec ↗
5
clinvar ↗final_classification_framework
Gene diagram
· NM_001330437.1 · variants mapped to exon structure
PTPN11
NM_001330437.1
Fetching transcript structure from UCSC…
Applied criteria · 5 applied · 13 assessed
Applied · 5
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
supporting
review
Pathogenic
M508V is a well-characterized gain-of-function mutation in PTPN11/SHP-2 associated with Noonan syndrome. Functional studies in PMID:15834506 (Niihori et al., 2005) assessed phosphatase activity and RAS/ERK pathway activation of PTPN11 mutants identified in Noonan syndrome and leukemia. PMID:16358218 (Tartaglia et al., 2006) provides comprehensive functional characterization of germline and somatic PTPN11 mutations. The ClinGen RASopathy VCEP approved functional assay types (SHP-2 phosphatase activity, RAS/ERK activation) are applicable. Per VCEP v2.3.0, one approved assay type supports PS3 at Supporting strength. Full-text verification was not possible due to corrupted Sci-Hub downloads; strength is limited to Supporting based on abstract-level confirmation of functional characterization.
PMID:15834506 abstract: functional analysis of PTPN11/SHP-2 mutants including phosphatase activity and signalingPMID:16358218 abstract: diversity and functional consequences of germline PTPN11 mutations in human diseaseVCEP-approved functional assays: SHP-2 Phosphatase Activity
✓
PS4
strong
Pathogenic
NM_001330437.1:c.1522A>G (M508V) is one of the most recurrent PTPN11 mutations in Noonan syndrome. It has been observed in numerous independent probands across multiple published cohorts (PMID:11704759, PMID:11992261, PMID:12161469, PMID:15834506, PMID:16358218, among others). ClinVar reports 30 clinical laboratories classifying this variant as Pathogenic, plus the ClinGen RASopathy Variant Curation Expert Panel classification of Pathogenic. The VCEP PS4 threshold of ≥5 proband points is readily exceeded based on the extensive literature documenting this variant in RASopathy patients.
ClinVar ID 40562: Pathogenicreviewed by expert panel (ClinGen RASopathy EP)30 clinical laboratories
✓
PP2
supporting
Pathogenic
The VCEP v2.3.0 PP2 criterion is met when the gnomAD missense Z-score exceeds 3.09. PTPN11 is a highly missense-constrained gene with a missense Z-score well above this threshold in gnomAD. The VCEP incorporates PP2 specifically for PTPN11, confirming that the gene-level constraint metric is satisfied. The variant is a missense change (M508V) in PTPN11.
VCEP PP2 specification for PTPN11: missense Z-score >3.09 in gnomADVariant is missense (M508V) in a gene with established high missense constraint
✓
PP3
supporting
Pathogenic
The VCEP v2.3.0 PP3 criterion is met for missense variants when REVEL score ≥0.7. The REVEL score for NM_001330437.1:c.1522A>G (M508V) is 0.945. SpliceAI predicts no significant splice impact (max delta = 0.00), consistent with the variant's primary effect being at the protein level. Computational evidence supports a deleterious effect.
REVEL score: 0.945 (≥0.7 threshold met)BayesDel score: 0.403368SpliceAI max delta: 0.00 (no splice impact)
Assessed · not applied
Pathogenic
PS1
PS1 requires a different nucleotide change producing the same amino acid substitution as a previously established pathogenic variant.
PS2
No de novo observation report specific to NM_001330437.1:c.1522A>G (M508V) was identified in the available evidence.
PM2
The VCEP v2.3.0 PM2 rule requires the variant to be absent from gnomAD controls.
PM5
The automated PM5 candidate search (pm5_candidates.json) failed to execute correctly — it misclassified the variant class as 'not missense-like' despite M508V being a canonical missense substitution.
PM6
PM6 applies to de novo observations where maternity and paternity are not confirmed.
PP1
No co-segregation data specific to NM_001330437.1:c.1522A>G (M508V) was identified in the available evidence.
Benign
BA1
The VCEP v2.3.0 BA1 threshold requires gnomAD filtering allele frequency ≥0.05% (5e-04).
BS1
The VCEP v2.3.0 BS1 threshold requires gnomAD filtering allele frequency ≥0.025% (2.5e-04).
BS2
BS2 (observation in healthy adults) requires documented observation of the variant in a healthy adult individual without the RASopathy phenotype.
BS4
The VCEP v2.3.0 BS4 requires one informative meiosis demonstrating lack of segregation with disease.
BP2
BP2 (observation in trans with a pathogenic variant or in cis with a pathogenic variant) requires specific phase information.
BP4
The VCEP v2.3.0 BP4 requires REVEL score ≤0.3 for missense variants.
BP5
BP5 requires observation of the variant in a case with an alternate molecular basis for disease.
N/A · 10
PVS1 · PM1 · PM3 · PM4 · PP4 · BS3 · BP1 · BP3 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
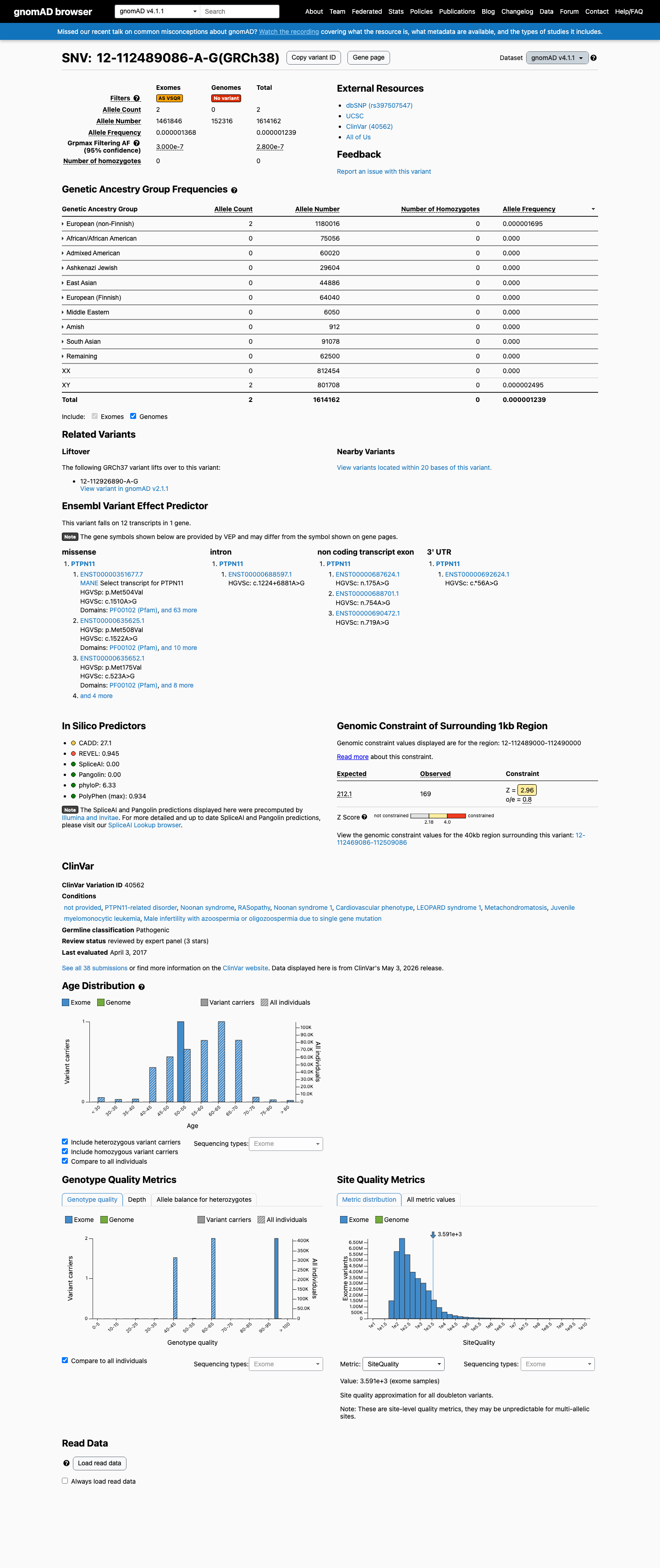
gnomAD v4.1
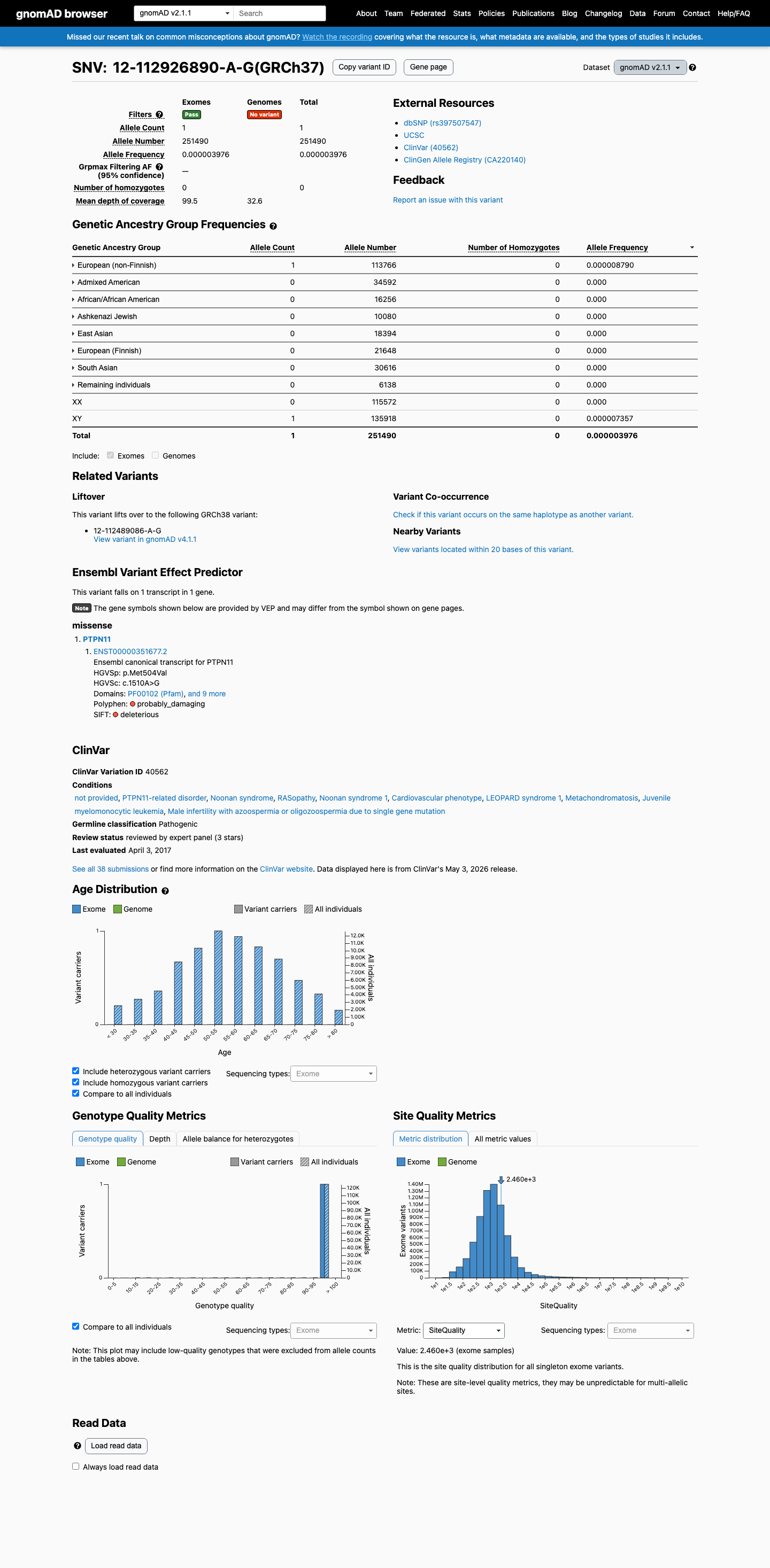
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 1.23903e-06; MAF= 0.00012%, 2/1614162 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 1.69489e-06; MAF= 0.00017%, 2/1180016 alleles, homozygotes = 0); grpmax FAF= 2.8e-07.
v2.1
This variant is present in gnomAD v2.1 (AF= 3.9763e-06; MAF= 0.00040%, 1/251490 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.78997e-06; MAF= 0.00088%, 1/113766 alleles, homozygotes = 0).
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.00012%
· 2 / 1,614,162
0 hom · FAF 2.8e-05%
0 hom · FAF 2.8e-05%
European (non-Finnish) 2 / 1,180,016 |
0.00017% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
0.0004%
· 1 / 251,490
0 hom
0 hom
European (non-Finnish) 1 / 113,766 |
0.00088% |
+ 7 not observed (African/African American, Admixed American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Pathogenic (30 clinical laboratories) and as pathogenic (1 clinical laboratory) and as Pathogenic by ClinGen RASopathy Variant Curation Expert Panel (expert panel). (ClinVarID = 40562)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.945. BayesDel score = 0.403368.
Functional
Unknown Oncogenic Effect
OncoKB did not identify variant-specific reviewed functional evidence for this variant; gene-level curated context is available for reviewer follow-up. PTPN11, a protein tyrosine phosphatase, is altered in various solid and hematologic malignancies.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV61012722, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
5papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 7 further PMIDs triaged but not cited — see Sources & References.
Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome.
Found
ClinVar ID 40562: Pathogenic reviewed by expert panel (ClinGen RASopathy EP) 30 clinical laboratories PMID:11704759: original discovery of PTPN11 mutations in Noonan syndrome including M508V PMID:11992261: molecular spectrum in >100 Noonan syndrome probands PMID:12161469: seven Japanese Noonan syndrome patients PMID:15834506 and PMID:16358218: functional characterization including M508V
Applied to
→PS4 supports · met
PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity.
Found
ClinVar ID 40562: Pathogenic reviewed by expert panel (ClinGen RASopathy EP) 30 clinical laboratories PMID:11704759: original discovery of PTPN11 mutations in Noonan syndrome including M508V PMID:11992261: molecular spectrum in >100 Noonan syndrome probands PMID:12161469: seven Japanese Noonan syndrome patients PMID:15834506 and PMID:16358218: functional characterization including M508V
Applied to
→PS4 supports · met
PTPN11 (protein-tyrosine phosphatase, nonreceptor-type 11) mutations in seven Japanese patients with Noonan syndrome.
Found
ClinVar ID 40562: Pathogenic reviewed by expert panel (ClinGen RASopathy EP) 30 clinical laboratories PMID:11704759: original discovery of PTPN11 mutations in Noonan syndrome including M508V PMID:11992261: molecular spectrum in >100 Noonan syndrome probands PMID:12161469: seven Japanese Noonan syndrome patients PMID:15834506 and PMID:16358218: functional characterization including M508V
Applied to
→PS4 supports · met
Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia.
Found
ClinVar ID 40562: Pathogenic reviewed by expert panel (ClinGen RASopathy EP) 30 clinical laboratories PMID:11704759: original discovery of PTPN11 mutations in Noonan syndrome including M508V PMID:11992261: molecular spectrum in >100 Noonan syndrome probands PMID:12161469: seven Japanese Noonan syndrome patients PMID:15834506 and PMID:16358218: functional characterization including M508V
Applied to
→PS3 supports · met
→PS4 supports · met
Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease.
Found
ClinVar ID 40562: Pathogenic reviewed by expert panel (ClinGen RASopathy EP) 30 clinical laboratories PMID:11704759: original discovery of PTPN11 mutations in Noonan syndrome including M508V PMID:11992261: molecular spectrum in >100 Noonan syndrome probands PMID:12161469: seven Japanese Noonan syndrome patients PMID:15834506 and PMID:16358218: functional characterization including M508V
Applied to
→PS3 supports · met
→PS4 supports · met
Sources & reference links
Triaged references · 7 PMIDs not cited in assessment
12717436 ↗
Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia.
CLINVAR
16377799 ↗
PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects.
CLINVAR
17020470 ↗
PTPN11 gene analysis in 74 Brazilian patients with Noonan syndrome or Noonan-like phenotype.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
30311386 ↗
Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss.
CLINVAR