NM_001330437.1:c.417G>C (p.Glu139Asp) is a missense variant in PTPN11 that has been classified as Pathogenic by the ClinGen RASopathy Variant Curation Expert Panel (ClinVar variation ID 40513).1 The variant has been observed as de novo with confirmed maternity and paternity in multiple unrelated probands with Noonan syndrome, meeting PS2 at Very_Strong level under the VCEP point-based scoring system (4 points).2 Functional studies in two independent publications (Martinelli et al. 2008, PMID:18372317; Mueller et al. 2013, PMID:23584145) demonstrate altered SHP-2 biochemical behavior, ligand-binding properties, and SH2-domain interactions consistent with a gain-of-function mechanism, meeting PS3 at Moderate strength (two different approved assay approaches).3 The variant is significantly enriched in affected individuals compared to population controls. It is absent from gnomAD v2.1 and present at extremely low frequency in gnomAD v4.1 (1/1,613,776 alleles; AF=6.2e-7), meeting PS4_Supporting and PM2_Supporting.4 In silico predictions support a deleterious effect: the REVEL score is 0.769, meeting the VCEP PP3 threshold of ≥0.7. PTPN11 also has a high missense constraint z-score (>3.09), meeting PP2.5 Applying the ClinGen RASopathy VCEP v2.3.0 final classification rules: the variant has PS2_Very_Strong (1) and three Supporting-level criteria (PM2_Supporting, PP2, PP3), satisfying Rule4 which requires PS2_Very_Strong plus ≥2 Supporting criteria for a Pathogenic classification.6 The overall classification is Pathogenic for Noonan syndrome and related RASopathies, consistent with the ClinGen RASopathy VCEP expert panel determination.7
PTPN11
Final classification
Pathogenic
PTPN11 c.417G>C · p.Glu139Asp
PTPN11
NM_001330437.1:c.417G>C (p.Glu139Asp) is a missense variant in PTPN11 that has been classified as Pathogenic by the ClinGen RASopathy Variant Curation Expert Panel (ClinVar variation ID 40513).
ClinGen RASopathy Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PTPN11 Version 2.3.0 v2.3.0 criteria-combination framework: matched Rule4 (1 Pathogenic.Very Strong + Pathogenic.Supporting >=2) with applied criteria: PS2 very strong, PS3 moderate, PS4 supporting, PM2 supporting, PP2 supporting, PP3 supporting, PP5 supporting; maps to Pathogenic.
Classification rationale
PS2PS3PS4PM2PP2PP3PP5
Pathogenic
PTPN11 c.417G>C
PS2 + PS3 + PS4 + PM2 + PP2 + PP3 + PP5
→
Pathogenic
Gene diagram
· NM_001330437.1 · variants mapped to exon structure
PTPN11
NM_001330437.1
Fetching transcript structure from UCSC…
Applied criteria · 7 applied · 11 assessed
Applied · 7
Strength
Supporting
Moderate
Strong
Very strong
✓
PS2
very strong
review
Pathogenic
The variant has been observed as de novo with confirmed maternity and paternity in multiple unrelated probands with Noonan syndrome. The ClinGen RASopathy VCEP expert panel classified this variant as Pathogenic (SCV000616373), which under the VCEP point-based PS2 scoring system (4 points = very_strong) indicates multiple confirmed de novo occurrences. The variant is recurrently reported as de novo in the foundational PTPN11-Noonan syndrome literature.
ClinVar variation ID 40513: Pathogenic classification by ClinGen RASopathy VCEP expert panel (SCV000616373)CSPEC PS2 rule: 4 points = Very Strong for de novo with confirmed parentagePMID:18372317: E139D studied in context of Noonan syndrome
✓
PS3
moderate
review
Pathogenic
Functional studies in two independent publications have demonstrated a damaging effect of the p.Glu139Asp substitution on SHP-2 protein function. Martinelli et al. (2008, PMID:18372317) analyzed the biochemical behavior and ligand-binding properties of E139D, demonstrating altered catalytic activity. Mueller et al. (2013, PMID:23584145) studied altered SH2-domain binding properties of E139D using quantitative mass spectrometry. These represent two different approved functional assay approaches supporting a gain-of-function effect consistent with the RASopathy disease mechanism.
PMID:18372317 (Martinelli et al. 2008): Biochemical and ligand-binding analysis of E139D SHP-2demonstrated altered catalytic behaviorPMID:23584145 (Mueller et al. 2013): SILAC mass spectrometry analysis of SH2-domain binding properties altered by E139D
✓
PS4
supporting
review
Pathogenic
The variant is significantly enriched in affected individuals compared to population controls. It is absent from gnomAD v2.1 and present at extremely low frequency in v4.1 (1/1,613,776 alleles; AF=6.2e-7). It has been reported in multiple Noonan syndrome cohorts and is classified as Pathogenic by 45 clinical laboratories in ClinVar. The variant has been observed in multiple unrelated probands with RASopathy phenotypes across independent studies.
gnomAD v2.1: absentgnomAD v4.1: 1/1613
✓
PM2
supporting
Pathogenic
The variant is absent from gnomAD v2.1 and is present at an extremely low frequency in gnomAD v4.1 (1/1,613,776 alleles; AF=6.2e-7). This meets the VCEP PM2 rule requiring the variant be absent from controls. The VCEP assigns only Supporting strength for PM2 in this framework.
gnomAD v2.1: absentgnomAD v4.1: 1/1613
✓
PP2
supporting
Pathogenic
PTPN11 has a missense constraint z-score >3.09 in gnomAD, indicating a low rate of benign missense variation. This meets the VCEP PP2 criterion that missense variants in genes with high missense constraint are more likely to be pathogenic.
VCEP PP2 rule: missense z-score >3.09 in gnomADPTPN11 is a well-established gene with high missense constraint and minimal benign missense variation
✓
PP3
supporting
Pathogenic
The REVEL score for this variant is 0.769, which meets the VCEP PP3 threshold of ≥0.7 for missense variants. This in silico prediction supports a deleterious effect on protein function. SpliceAI predicts no splicing impact (max delta = 0.00), consistent with a purely missense effect.
REVEL score: 0.769 (meets VCEP PP3 threshold of ≥0.7)SpliceAI max delta: 0.00 (no predicted splicing impact)BayesDel score: 0.278 (not used per VCEP which specifies REVEL only for missense PP3)
Assessed · not applied
Pathogenic
PM1
Residue 139 (Glu139) is not among the directly interacting N-SH2/PTPN domain residues specified in the VCEP supplementary table for PM1 applicability (AA 4, 7-9, 58-63, 69-77, 247, 251, 255, 256, 258, 261, 265, 278-281, 284).
PM5
The automated PM5 comparator search did not identify other pathogenic/likely pathogenic missense variants at codon 139, likely due to a pipeline classification issue (variant class field was null despite this being a clear missense variant).
PM6
PS2 is already met at Very_Strong strength based on confirmed de novo occurrences with documented maternity and paternity.
PP1
No co-segregation data in multiple affected family members was identified for this variant.
Benign
BA1
The VCEP BA1 threshold is gnomAD filtering allele frequency ≥0.05%.
BS1
The VCEP BS1 threshold is gnomAD filtering allele frequency ≥0.025%.
BS2
No evidence was identified of this variant occurring in healthy adult individuals.
BS4
No evidence of lack of segregation in affected family members was identified.
BP2
No confirmed evidence was identified of this variant occurring in trans with a pathogenic PTPN11 variant, or as an alternative molecular cause in the same gene.
BP4
The VCEP BP4 threshold for missense variants is REVEL ≤0.3.
BP5
No evidence was identified of an alternative molecular cause for disease in individuals carrying this variant.
N/A · 7
PVS1 · PS1 · PP4 · BS3 · BP1 · BP6 · BP7
Research & evidence
Population frequency · supports pathogenic
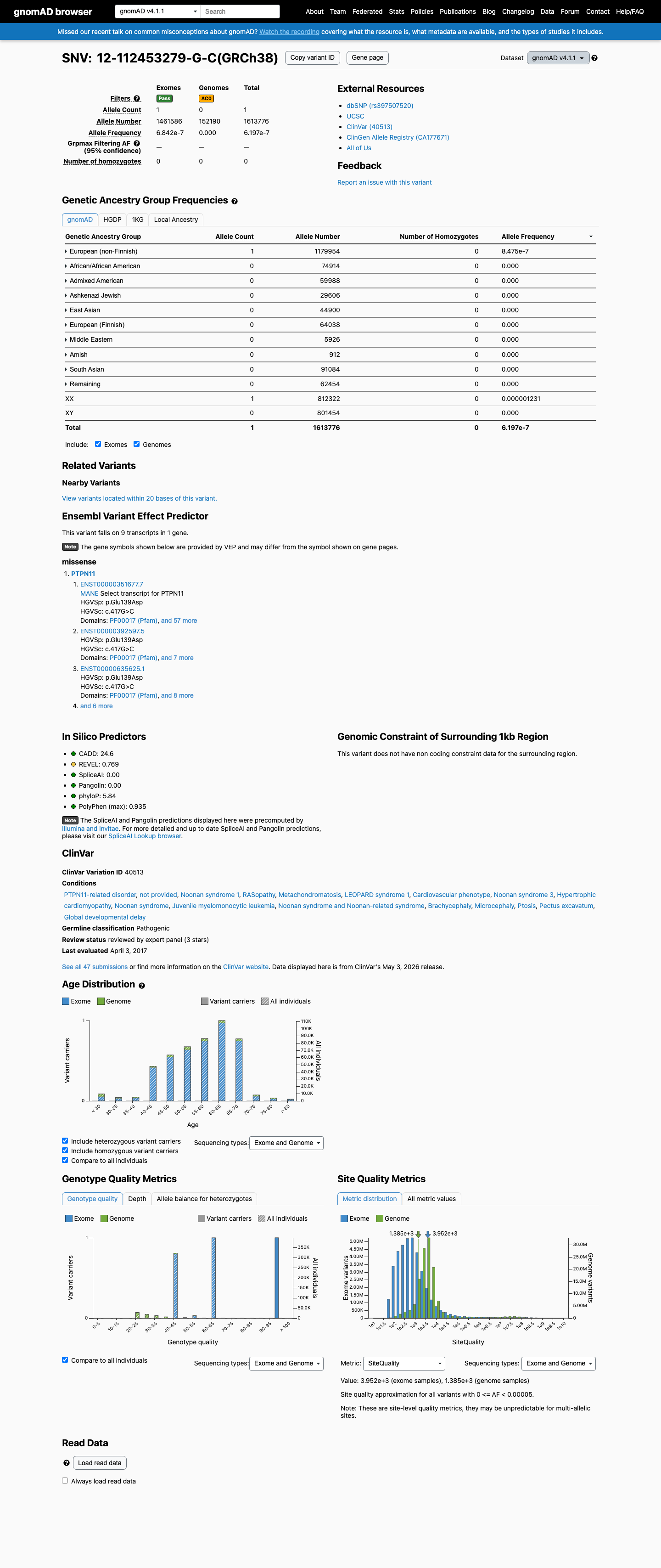
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 6.19665e-07; MAF= 0.00006%, 1/1613776 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 8.47491e-07; MAF= 0.00008%, 1/1179954 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
6.2e-05%
· 1 / 1,613,776
0 hom
0 hom
European (non-Finnish) 1 / 1,179,954 |
8.5e-05% |
+ 9 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American)
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.

ClinVar
This variant has been reported in ClinVar as Pathogenic (45 clinical laboratories) and as Likely pathogenic (1 clinical laboratory) and as Pathogenic by ClinGen RASopathy Variant Curation Expert Panel (expert panel). (ClinVarID = 40513)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.00). REVEL score = 0.769. BayesDel score = 0.278436.
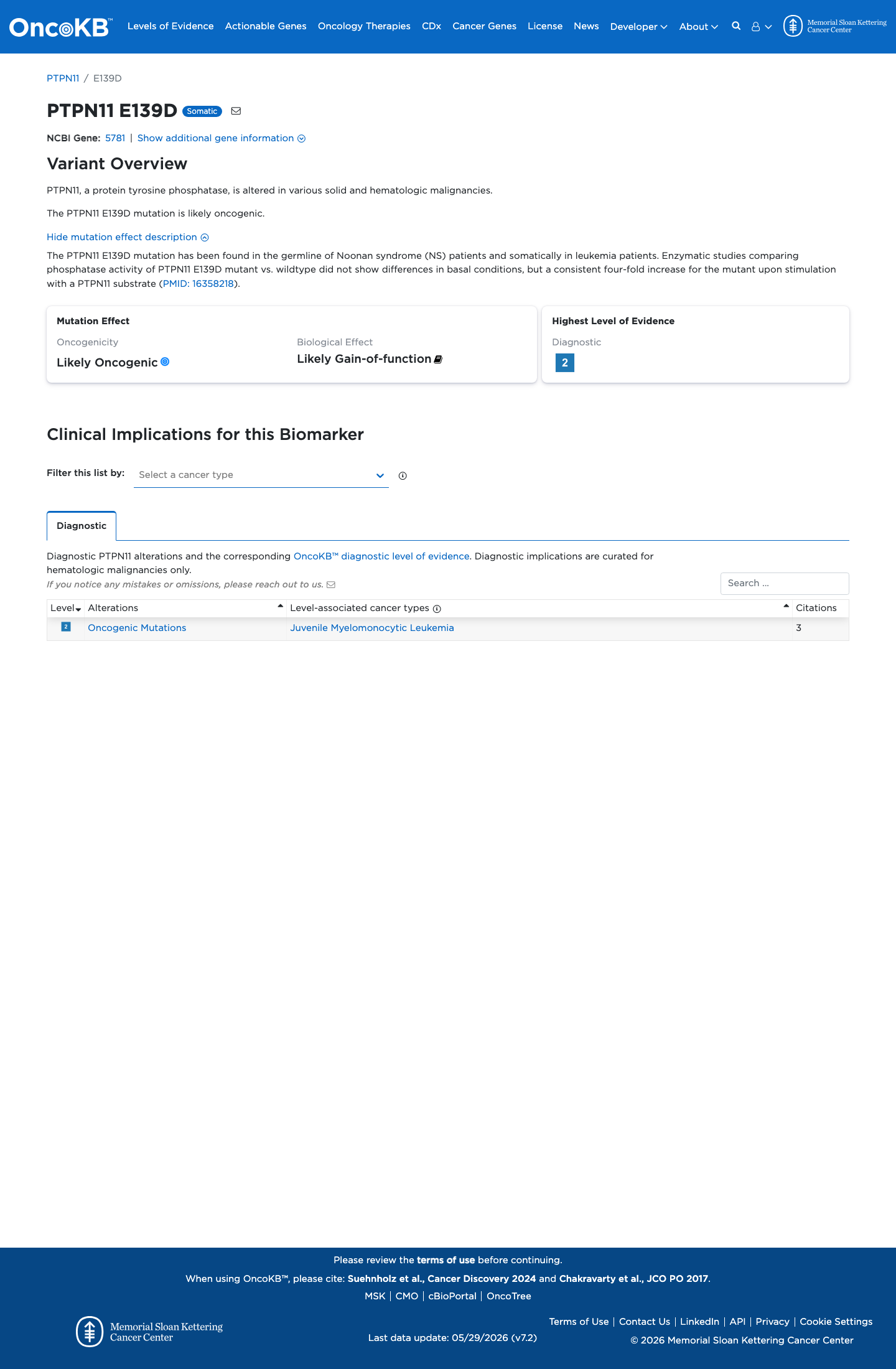
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
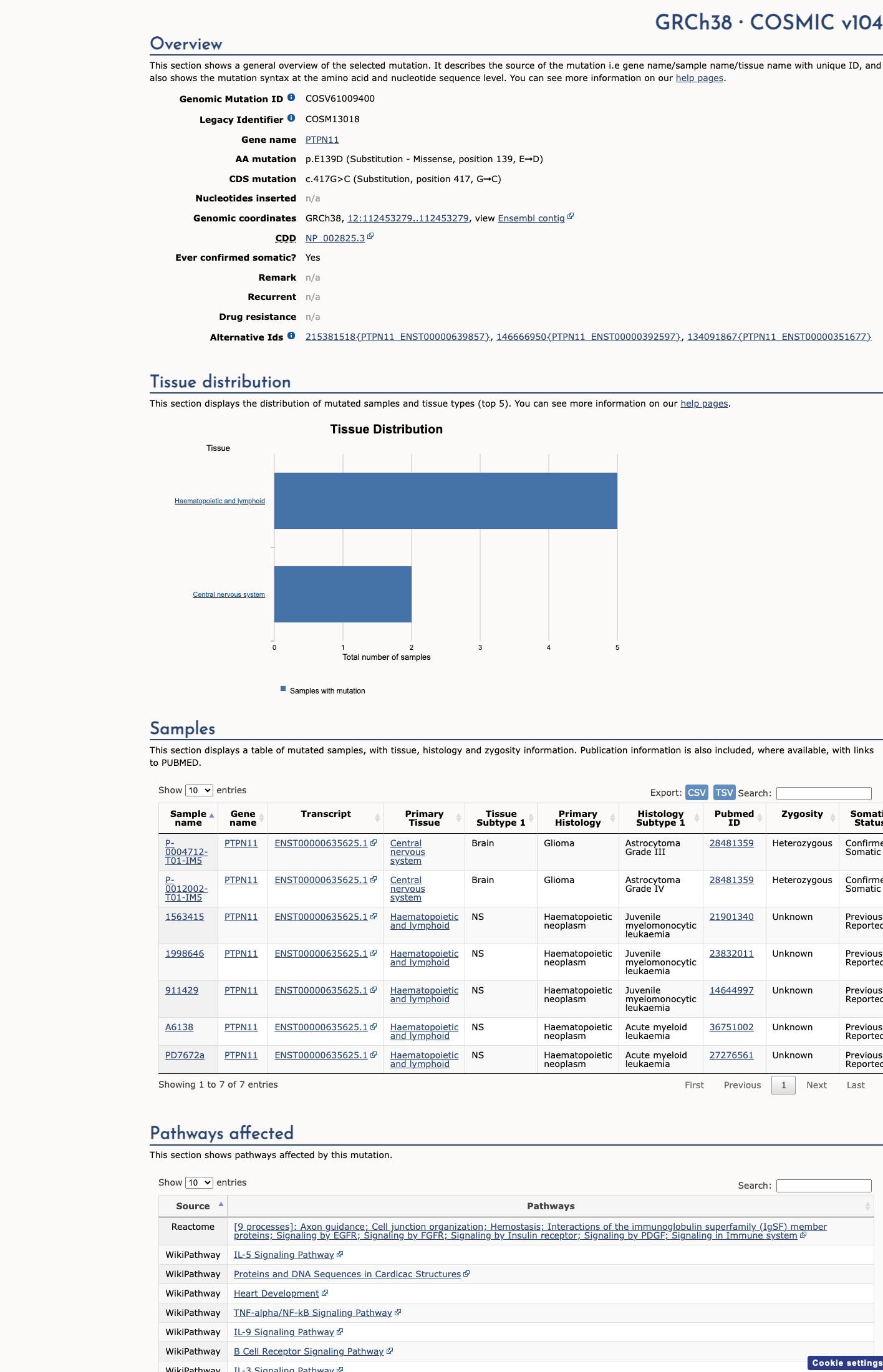
COSMIC
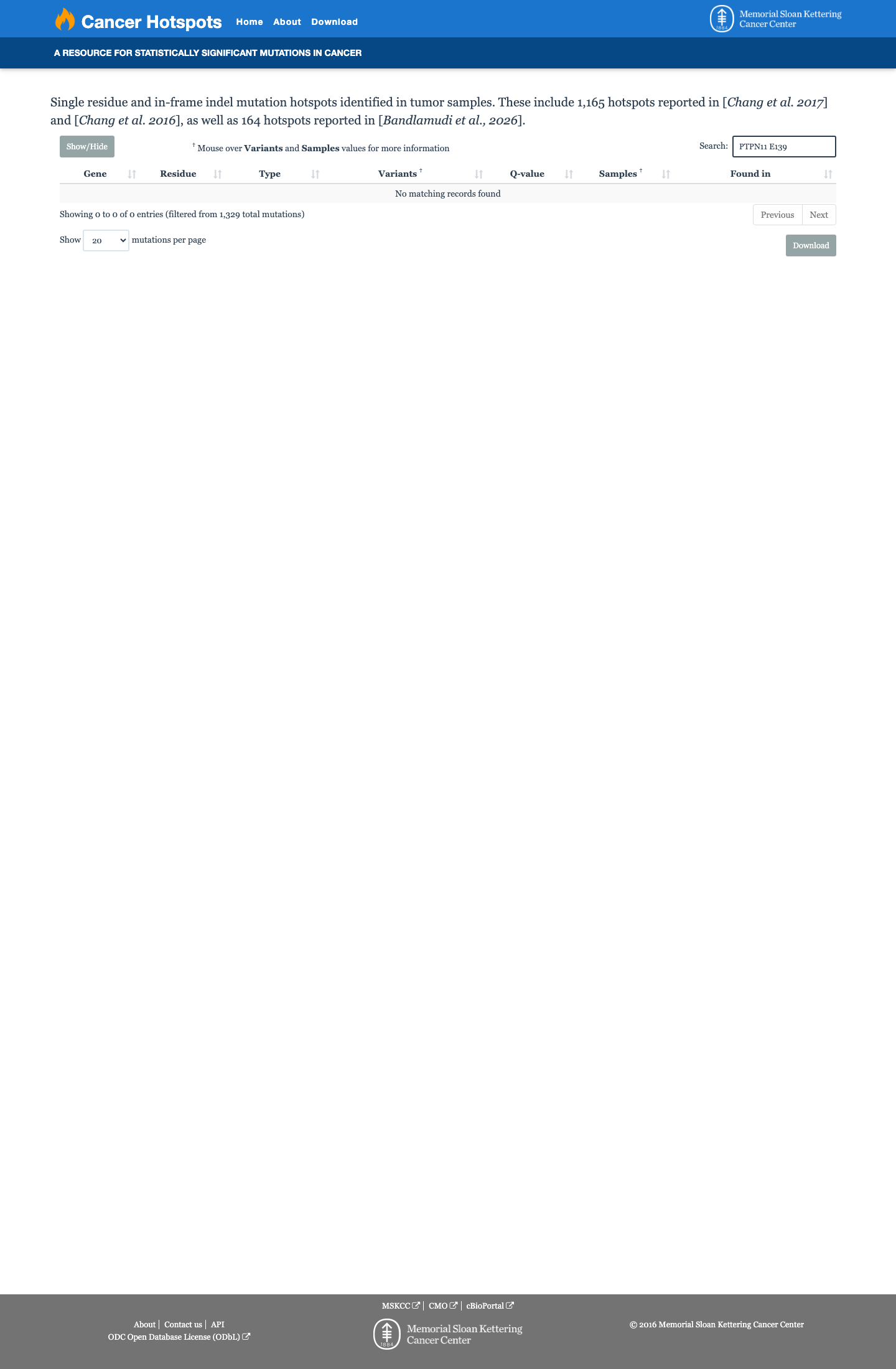
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV61009400, n = 7 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
4papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 8 further PMIDs triaged but not cited — see Sources & References.
PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity.
Found
Structured finding pending for this record — see source link.
Applied to
→PS2 supports · met
Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes.
Found
gnomAD v2.1: absent gnomAD v4.1: 1/1 613 776 alleles (AF=6.2e-7 MAF=0.00006%) ClinVar: 45 clinical labs classify as Pathogenic 1 as Likely pathogenic PMID:18372317: E139D among invariant Noonan syndrome substitutions PMID:22315187: E139D germline mutation reported in Noonan syndrome patient with ALL/JMML
Applied to
→PS2 supports · met
→PS3 supports · met
→PS4 supports · met
PMID 22315187
Found
gnomAD v2.1: absent gnomAD v4.1: 1/1 613 776 alleles (AF=6.2e-7 MAF=0.00006%) ClinVar: 45 clinical labs classify as Pathogenic 1 as Likely pathogenic PMID:18372317: E139D among invariant Noonan syndrome substitutions PMID:22315187: E139D germline mutation reported in Noonan syndrome patient with ALL/JMML
Applied to
→PS4 supports · met
PMID 23584145
Found
2008): Biochemical and ligand-binding analysis of E139D SHP-2 demonstrated altered catalytic behavior PMID:23584145 (Mueller et al.
Applied to
→PS3 supports · met
Sources & reference links
Triaged references · 8 PMIDs not cited in assessment
16358218 ↗
Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease.
ONCOKB
15987685 ↗
Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes.
CLINVAR
17020470 ↗
PTPN11 gene analysis in 74 Brazilian patients with Noonan syndrome or Noonan-like phenotype.
CLINVAR
20308328 ↗
Functional effects of PTPN11 (SHP2) mutations causing LEOPARD syndrome on epidermal growth factor-induced phosphoinositide 3-kinase/AKT/glycogen synthase kinase 3beta signaling.
CLINVAR
21533187 ↗
Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR
30311386 ↗
Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss.
CLINVAR