The variant NM_001354609.1:c.1068A>G (p.Gln356=) is a synonymous substitution in BRAF exon 8, outside the critical functional domains defined by the ClinGen RASopathy VCEP.1 This variant is present in gnomAD v2.1 at an allele frequency of 0.02546% (72/282,848 alleles, 1 homozygote) with a grpmax filtering allele frequency of 0.1388%, and in gnomAD v4.1 at 0.01394% (225/1,614,110 alleles, 2 homozygotes) with a grpmax FAF of 0.1350%.2 BA1 is met at stand-alone benign strength: the gnomAD grpmax FAF of 0.1388% (v2.1) and 0.1350% (v4.1) exceeds the CSPEC RASopathy VCEP threshold of ≥0.05%.3 BS1 is met at strong benign strength: the gnomAD grpmax FAF exceeds the CSPEC RASopathy VCEP threshold of ≥0.025%.4 BS2 is met at strong level: homozygous individuals are observed in gnomAD (1 in v2.1, 2 in v4.1), which is inconsistent with a highly penetrant autosomal dominant RASopathy disorder.5 BP4 is met at supporting benign strength: SpliceAI predicts no significant splice impact (max delta 0.01), satisfying the VCEP BP4 criterion for negligible predicted splicing outcome.6 BP7 is met at supporting benign strength: the variant is synonymous (p.Gln356=) with no predicted splice impact (SpliceAI delta 0.01), applied in conjunction with BP4.7 The ClinGen RASopathy Variant Curation Expert Panel has classified this variant as Benign (ClinVar Variation ID 44788), applying criteria BA1, BS1, BP4, and BP7, consistent with this independent assessment.8 Under the CSPEC RASopathy VCEP v2.3.0 classification framework Rule 17, BA1 alone (stand-alone benign) is sufficient for a Benign classification. Additionally, BS1 (strong) and BS2 (strong) satisfy Rule 16 (≥2 strong benign criteria). The variant is classified as Benign.9
BRAF
Final classification
Benign
BRAF c.1068A>G · p.Gln356=
BRAF
The variant NM_001354609.1:c.1068A>G (p.Gln356=) is a synonymous substitution in BRAF exon 8, outside the critical functional domains defined by the ClinGen RASopathy VCEP.
ClinGen RASopathy Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for BRAF Version 2.3.0 v2.3.0 criteria-combination framework: matched Rule17 (1 Benign.Stand Alone) with applied criteria: BA1 stand-alone benign, BS1 strong benign, BS2 strong benign, BP4 supporting benign, BP6 supporting benign, BP7 supporting benign; maps to Benign.
Classification rationale
BA1BS1BS2BP4BP6BP7
Benign
BRAF c.1068A>G
BA1 + BS1 + BS2 + BP4 + BP6 + BP7
→
Benign
Gene diagram
· NM_001354609.1 · variants mapped to exon structure
BRAF
NM_001354609.1
Fetching transcript structure from UCSC…
Applied criteria · 6 applied · 12 assessed
Applied · 6
Strength
Supporting
Moderate
Strong
Very strong
✓
BA1
stand-alone
Benign
The variant has a gnomAD filtering allele frequency (grpmax FAF) of 0.1388% in v2.1 and 0.1350% in v4.1, exceeding the CSPEC RASopathy VCEP BA1 threshold of ≥0.05%. This is consistent with a benign population polymorphism for a dominant RASopathy disorder.
gnomAD v2.1 grpmax FAF 0.1388% ≥ 0.05% thresholdgnomAD v4.1 grpmax FAF 0.1350% ≥ 0.05% thresholdCSPEC BA1 rule satisfied
✓
BS1
strong
Benign
The variant has a gnomAD filtering allele frequency (grpmax FAF) of 0.1388% in v2.1 and 0.1350% in v4.1, exceeding the CSPEC RASopathy VCEP BS1 threshold of ≥0.025%. The observed frequency in the general population is inconsistent with a highly penetrant dominant RASopathy disorder.
gnomAD v2.1 grpmax FAF 0.1388% ≥ 0.025% thresholdgnomAD v4.1 grpmax FAF 0.1350% ≥ 0.025% thresholdCSPEC BS1 rule satisfied
✓
BS2
strong
Benign
Homozygotes for this variant are observed in gnomAD (1 in v2.1, 2 in v4.1). For a dominant disorder such as RASopathy with full penetrance expected at an early age, observation of homozygous healthy individuals in population databases constitutes strong evidence for a benign classification.
gnomAD v2.1: 1 homozygote (72 total alleles)gnomAD v4.1: 2 homozygotes (225 total alleles)homozygotes in presumably healthy population controls for an autosomal dominant disorder
✓
BP4
supporting
Benign
CSPEC RASopathy VCEP BP4 for splicing variants requires predicted outcome to be negligible or not matching disease mechanism. SpliceAI predicts no significant splice impact for this synonymous variant (max delta score 0.01), satisfying the BP4 criteria for a negligible predicted splicing outcome.
SpliceAI max delta 0.01no predicted splicing impactsatisfies VCEP BP4 for negligible splicing outcome
✓
✓
BP7
supporting
Benign
This is a synonymous (silent) variant (p.Gln356=) for which SpliceAI predicts no impact to the splice consensus sequence nor creation of a new splice site (max delta 0.01). The nucleotide is not in a known highly conserved functional element within the critical domains defined by the VCEP. BP7 is applied in conjunction with BP4 per CSPEC guidance.
Synonymous variant p.Gln356=SpliceAI delta 0.01 (no splice impact)applied in conjunction with BP4 per VCEP BP7 guidance
Assessed · not applied
Pathogenic
PS1
PS1 requires the same amino acid change as a previously established pathogenic variant.
PS2
No confirmed de novo occurrences (with maternity and paternity confirmation) have been reported in RASopathy patients for this variant.
PS3
No dedicated in vitro functional assay data (e.g., minigene splicing, kinase activity) has been identified for this exact variant.
PS4
The variant is present in gnomAD at population frequencies (v2.1: 0.02546%, 72/282,848 alleles; v4.1: 0.01394%, 225/1,614,110 alleles) that far exceed any plausible enrichment in RASopathy cases.
PM1
CSPEC RASopathy VCEP restricts PM1 to critical and well-established functional domains: exon 6, exon 11, P-loop (AA 459-474), and CR3 activation segment (AA 594-627).
PM2
CSPEC RASopathy VCEP requires the variant to be absent from controls (gnomAD) for PM2_Supporting.
PM6
No assumed de novo occurrences (without maternity/paternity confirmation) have been reported for this variant in RASopathy probands.
PP1
No published multigenerational families demonstrating cosegregation of this variant with RASopathy in multiple affected relatives.
PP3
CSPEC RASopathy VCEP PP3 for missense variants requires REVEL ≥ 0.7.
Benign
BS4
No published segregation data (lack of cosegregation with disease in affected families) has been identified for this variant.
BP2
No evidence has been identified of this variant occurring in trans with a known pathogenic BRAF variant in a RASopathy patient.
BP5
The CSPEC RASopathy VCEP BP5 criterion uses a point-based system.
N/A · 7
PVS1 · PM5 · PP2 · PP4 · PP5 · BS3 · BP1
Research & evidence
Population frequency · supports benign
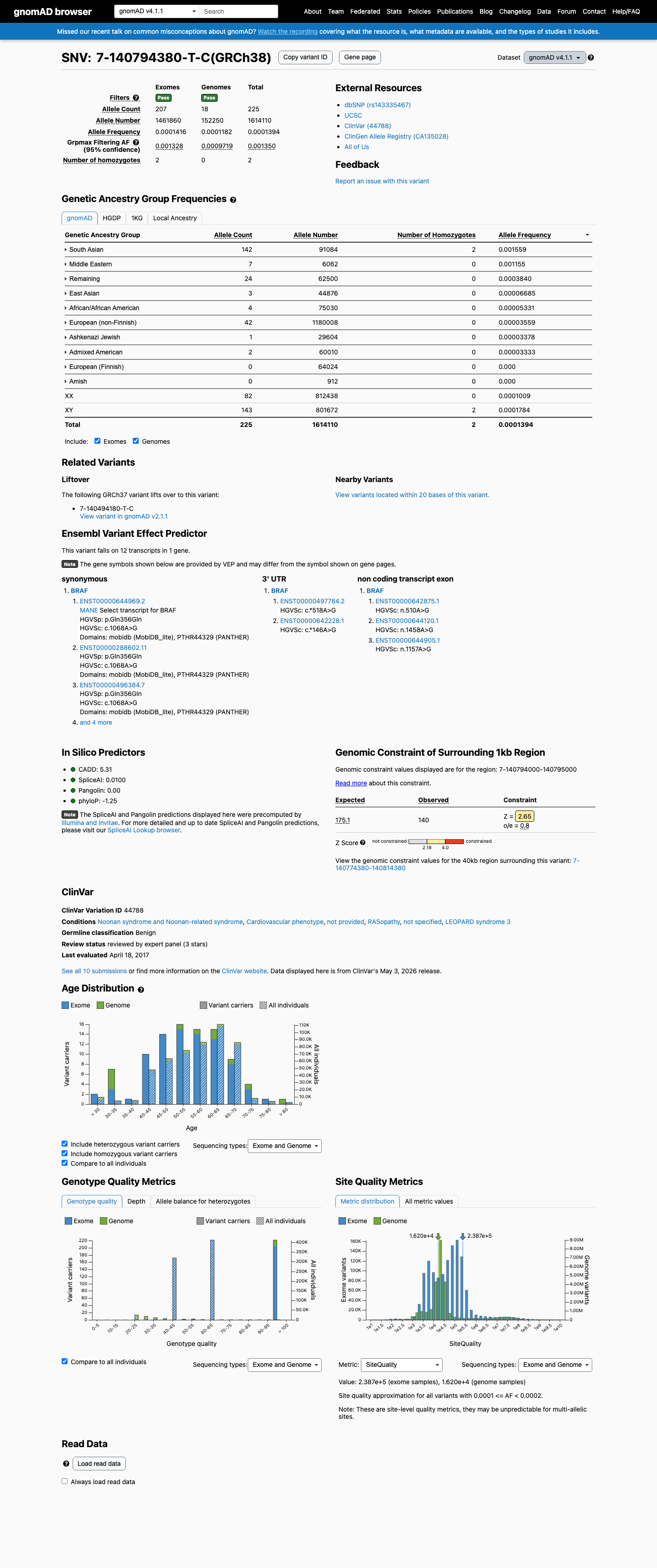
gnomAD v4.1
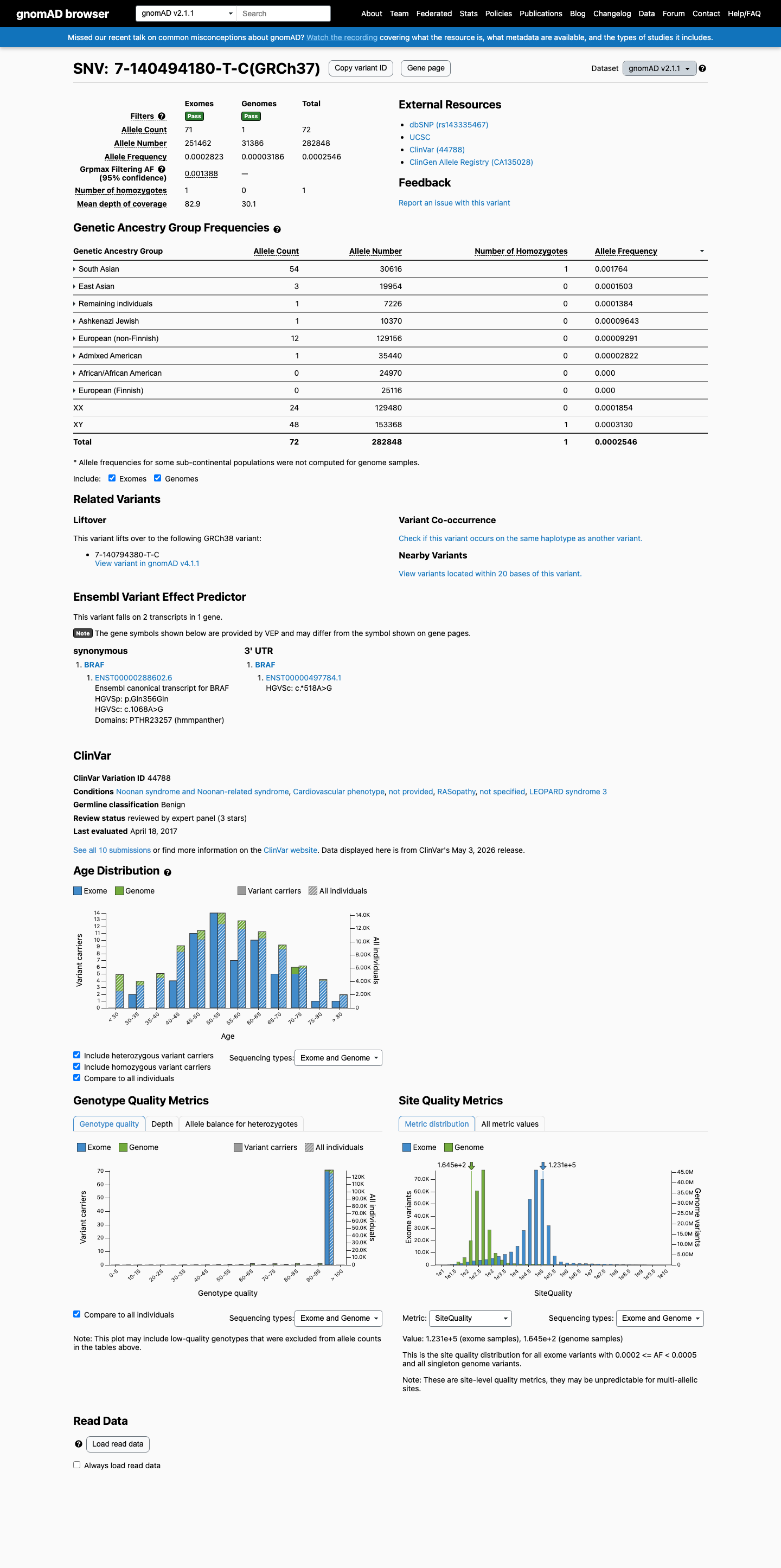
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0.000139396; MAF= 0.01394%, 225/1614110 alleles, homozygotes = 2) and has highest observed frequency in the South Asian population (AF= 0.001559; MAF= 0.15590%, 142/91084 alleles, homozygotes = 2); grpmax FAF= 0.00134985.
v2.1
This variant is present in gnomAD v2.1 (AF= 0.000254554; MAF= 0.02546%, 72/282848 alleles, homozygotes = 1) and has highest observed frequency in the South Asian population (AF= 0.00176378; MAF= 0.17638%, 54/30616 alleles, homozygotes = 1); grpmax FAF= 0.00138788.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.014%
· 225 / 1,614,110
2 hom · FAF 0.13%
2 hom · FAF 0.13%
South Asian 142 / 91,084 |
0.16% 2 hom |
Middle Eastern 7 / 6,062 |
0.12% |
Remaining individuals 24 / 62,500 |
0.038% |
East Asian 3 / 44,876 |
0.0067% |
African/African American 4 / 75,030 |
0.0053% |
European (non-Finnish) 42 / 1,180,008 |
0.0036% |
Ashkenazi Jewish 1 / 29,604 |
0.0034% |
Admixed American 2 / 60,010 |
0.0033% |
+ 2 not observed (European (Finnish), Amish)
gnomAD v2.1
0.025%
· 72 / 282,848
1 hom · FAF 0.14%
1 hom · FAF 0.14%
South Asian 54 / 30,616 |
0.18% 1 hom |
East Asian 3 / 19,954 |
0.015% |
Remaining individuals 1 / 7,226 |
0.014% |
Ashkenazi Jewish 1 / 10,370 |
0.0096% |
European (non-Finnish) 12 / 129,156 |
0.0093% |
Admixed American 1 / 35,440 |
0.0028% |
+ 2 not observed (African/African American, European (Finnish))
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
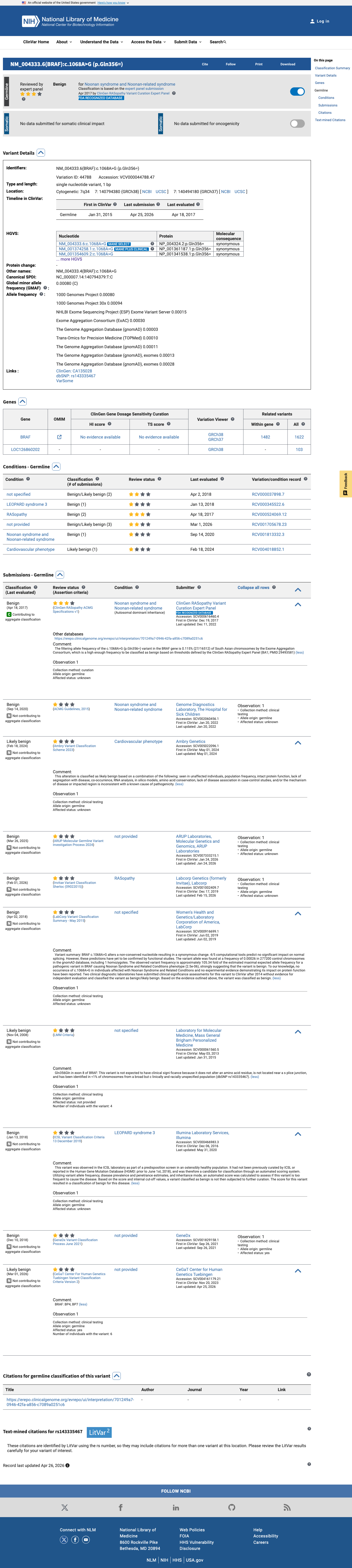
ClinVar
This variant has been reported in ClinVar as Benign (6 clinical laboratories) and as Likely benign (3 clinical laboratories) and as Benign by ClinGen RASopathy Variant Curation Expert Panel (expert panel). (ClinVarID = 44788)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01).
Functional
Unknown Oncogenic Effect
OncoKB identified curated literature and non-variant-specific oncogenicity context for review; listed oncogenicity label: Unknown Oncogenic Effect.
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has not previously been reported in somatic cancers (COSMIC).
Hotspots
This variant does not lie in a statistically significant hotspot.
Sources & reference links
Triaged references · 4 PMIDs not cited in assessment
24033266 ↗
A systematic approach to assessing the clinical significance of genetic variants.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR