Classification rationale
BP4BP6BS1BA1
Benign
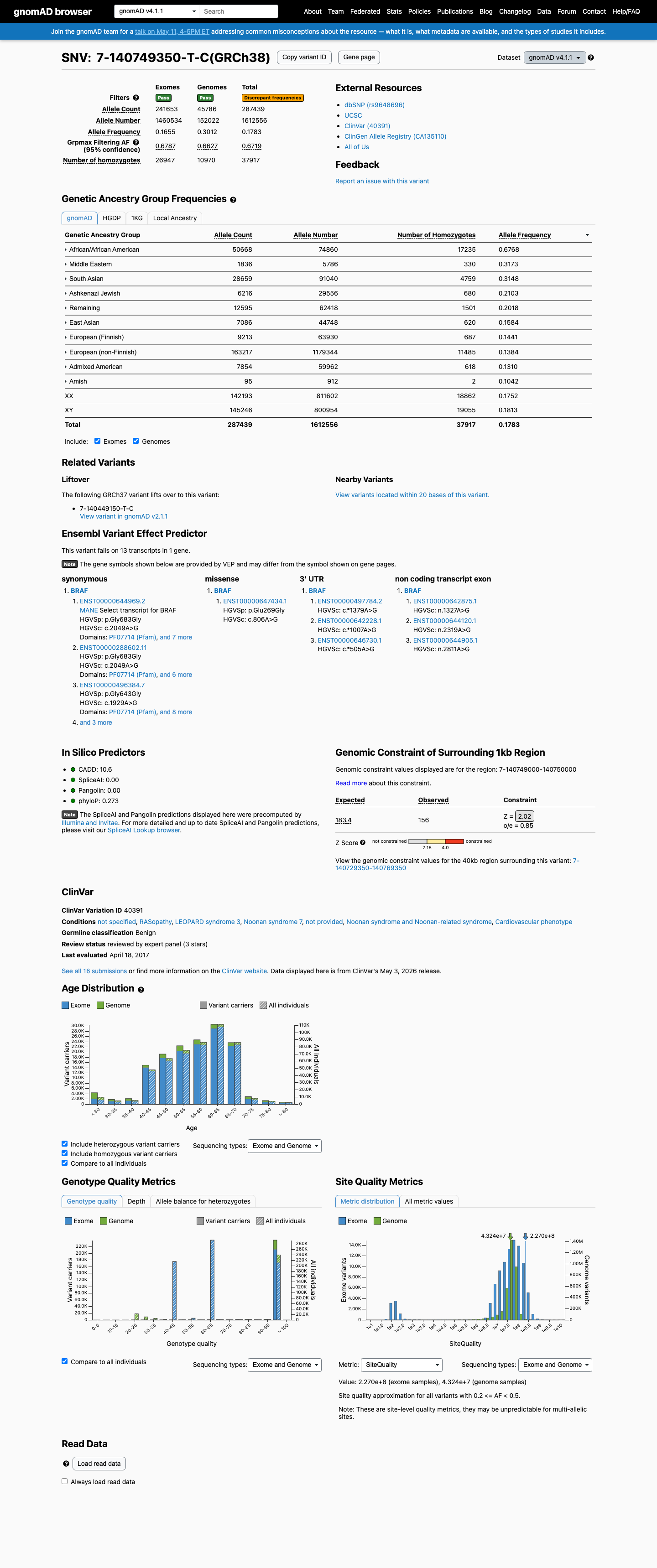
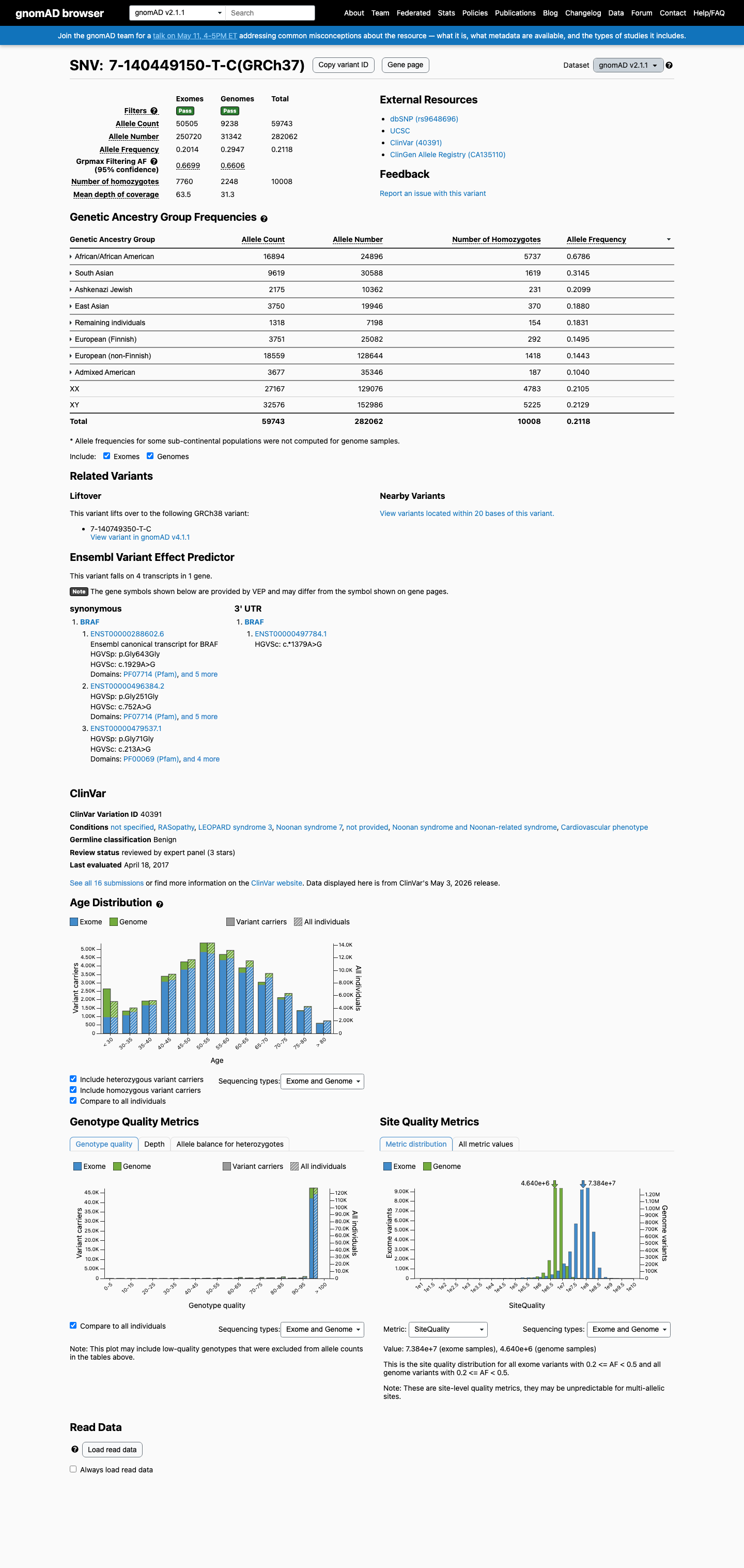
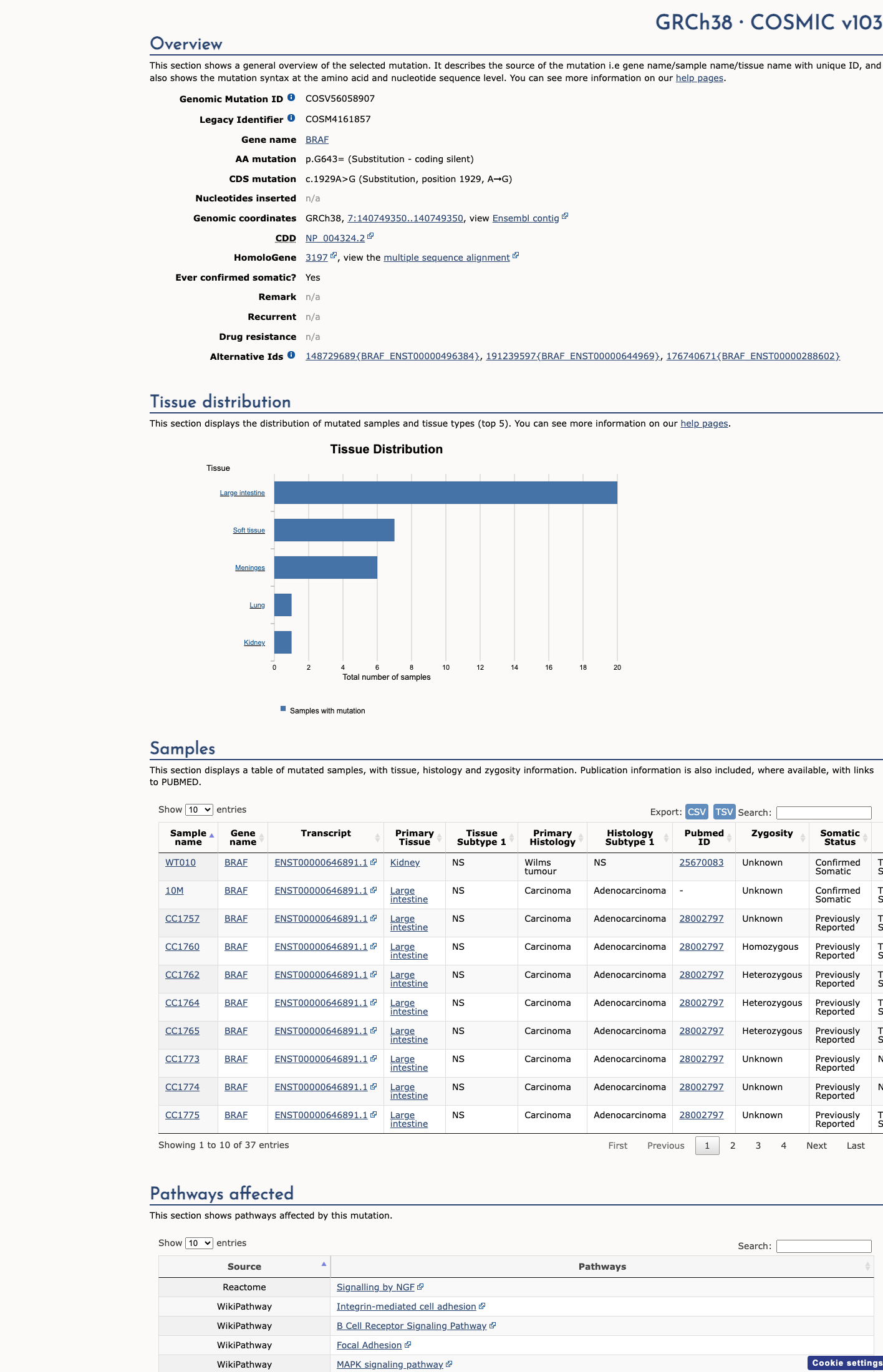
BRAF c.1929A>G
The BRAF c.1929A>G (p.Gly643=) variant is reported in ClinVar as Benign with expert-panel review.1 This variant is common in population databases, with an allele frequency of 21.18080% in gnomAD v2.1 and 17.82506% in gnomAD v4.1, far above the RASopathy VCEP BA1 threshold of 0.05% and BS1 threshold of 0.025%.2 Computational evidence does not support a damaging effect, with a REVEL score of 0.247 and SpliceAI predicting no significant splice impact with a maximum delta score of 0.01.3
BP4 + BP6 + BS1 + BA1
→
Benign