NM_001412.4:c.26G>A (p.Gly9Asp) is a missense variant in exon 2 of EIF1AX, a gene encoding a translation initiation factor in which N-terminal tail (NTT) missense mutations are established oncogenic drivers.1 The variant is absent from gnomAD v2.1 and v4.1 (0/1,168,429 alleles), meeting PM2 (supporting).2 The p.Gly9 residue lies within the NTT functional domain (residues ~2-15), a well-characterized hotspot for gain-of-function cancer mutations that impair scanning arrest and enhance translation of cell cycle genes. This satisfies PM1 at moderate strength.3 Functional characterization by Sehrawat et al. (2019) demonstrated that the G9D/R13H double mutant reduces binding to ribosomal protein Rps10, and other NTT mutants including G9R (same position) enhance translation of long 5'UTR mRNAs. OncoKB independently curates this variant as Likely Oncogenic with Likely Gain-of-function effect. This satisfies PS3 at supporting strength.4 In silico predictors favor a neutral effect: BayesDel score is -0.034 (predicted benign) and SpliceAI max delta is 0.02 (no splice impact). This satisfies BP4 (supporting benign).5 The overall evidence profile consists of one moderate pathogenic criterion (PM1), two supporting pathogenic criteria (PM2, PS3), and one supporting benign criterion (BP4). Per the generic ACMG/AMP 2015 combination rules (PMID:25741868), one moderate plus two supporting criteria does not reach the threshold for Likely Pathogenic (which requires two moderate and two supporting, or three moderate, or one strong and two supporting). The single supporting benign criterion does not independently reach any benign classification threshold. The variant is therefore classified as a Variant of Uncertain Significance (VUS).6
EIF1AX
Final classification
VUS
EIF1AX c.26G>A · p.Gly9Asp
EIF1AX
NM_001412.4:c.26G>A (p.Gly9Asp) is a missense variant in exon 2 of EIF1AX, a gene encoding a translation initiation factor in which N-terminal tail (NTT) missense mutations are established oncogenic drivers.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PS3 supporting, PM1 moderate, PM2 supporting, BP4 supporting benign; combination = 1 moderate + 2 supporting + 1 supporting benign, which maps to VUS.
Classification rationale
PS3PM1PM2
BP4
VUS
EIF1AX c.26G>A
PS3 + PM1 + PM2 + BP4
→
VUS
5
bayesdelspliceai ↗
6
generic_acmg_combination_rules
Gene diagram
· NM_001412.4 · variants mapped to exon structure
EIF1AX
NM_001412.4
Fetching transcript structure from UCSC…
Applied criteria · 4 applied · 18 assessed
Applied · 4
Strength
Supporting
Moderate
Strong
Very strong
✓
PS3
supporting
review
Pathogenic
Functional characterization of the EIF1AX N-terminal tail (NTT) domain, where p.Gly9Asp resides, was performed by Sehrawat et al. (PMID:30420357). The G9D/R13H double mutant showed reduced binding to ribosomal protein Rps10 in vitro, consistent with impaired scanning arrest and gain-of-function. Additionally, G9R (same position, different substitution) and other NTT mutants (G6D, G8R, K10E) showed enhanced translation of long 5'UTR mRNAs. The exact G9D single mutant was generated but its standalone functional data were not separately reported; the G9D/R13H double mutant data and same-position G9R data are suggestive but not definitive for the isolated G9D variant. The somatic OncoKB curation classifies this variant as Likely Oncogenic with Likely Gain-of-function effect, providing additional orthogonal support.
G9D/R13H double mutant exhibited reduced Rps10 binding in in vitro pull-down assay (PMID:30420357Figure 8)G9R single mutant (same residue) and other NTT mutants showed enhanced scanning of long 5'UTR mRNAs (PMID:30420357
✓
PM1
moderate
Pathogenic
The variant p.Gly9Asp is located in the N-terminal tail (NTT) of EIF1AX (residues ~2-15), a well-characterized functional domain critical for translation initiation fidelity. The NTT is established as a hotspot for cancer-associated somatic mutations in uveal melanoma, thyroid cancer, and ovarian cancer (PMID:30420357, PMID:30305285). Cancerhotspots.org identifies this residue as statistically significant. Multiple publications confirm that NTT mutations confer gain-of-function by impairing scanning arrest and enhancing translation of cell cycle genes. Gly9 is among the most frequently mutated residues in the NTT domain.
NTT domain (residues 2-15) is a critical functional domain for translation initiation and scanning arrestCancerhotspots.org identifies p.Gly9 as a statistically significant hotspot residueMultiple publications (PMID:30420357
✓
PM2
supporting
Pathogenic
This variant is absent from large population databases. gnomAD v2.1: absent. gnomAD v4.1: 0 alleles out of 1,168,429 (AF = 0.0%). gnomAD-Canada v1.0: absent. The variant meets the PM2 threshold of <0.1% allele frequency in all populations.
gnomAD v2.1: absentgnomAD v4.1: 0/1168
✓
BP4
supporting
Benign
Multiple lines of computational evidence suggest no impact on the gene product. BayesDel score is -0.034 (negative scores predict a benign effect). SpliceAI max delta score is 0.02, predicting no splicing impact. REVEL and HCI prior are unavailable but the available in silico tools consistently support a neutral or benign prediction.
BayesDel score: -0.034 (predicted benign)SpliceAI max delta: 0.02 (no predicted splice impact)
Assessed · not applied
Pathogenic
PS1
No evidence was identified that a different nucleotide change at c.26 leading to the same p.Gly9Asp amino acid substitution has been classified as pathogenic in a clinical database or the literature.
PS2
No de novo data (parental testing results, trio sequencing) is available for this variant.
PS4
No case-control data or systematically ascertained affected vs.
PM5
No confirmed pathogenic missense variant at the same amino acid position (Gly9) was identified in ClinVar.
PM6
No de novo data or confirmed maternity/paternity information is available.
PP1
No cosegregation data with disease in multiple affected family members is available for this variant.
PP2
Insufficient data to determine whether EIF1AX has a low rate of benign missense variation and whether missense variants are a common disease mechanism in a germline context.
PP3
In silico tools do not support a damaging effect.
PP4
No proband phenotype or clinical data is available to evaluate phenotype specificity or gene-disease fit.
PP5
No reputable source has independently classified this variant as pathogenic for a germline disorder.
Benign
BA1
The variant allele frequency in gnomAD v4.1 is 0.0% (0/1,168,429 alleles), well below the BA1 threshold of >1%.
BS1
The variant allele frequency in gnomAD v4.1 is 0.0% (0/1,168,429 alleles), well below the BS1 threshold of >0.3%.
BS2
No data available regarding observation of this variant in healthy adult individuals with full penetrance expected.
BS3
Functional studies in the literature do not demonstrate a benign effect.
BS4
No family segregation data is available to evaluate whether the variant does not segregate with disease in affected family members.
BP2
No data available on observation of this variant in trans with a known pathogenic variant for a fully penetrant dominant disorder, or in cis with a pathogenic variant for a recessive disorder.
BP5
No alternative molecular basis for disease has been identified in the available data.
BP6
No reputable source has classified this variant as benign or likely benign.
N/A · 6
PVS1 · PM3 · PM4 · BP1 · BP3 · BP7
Research & evidence
Population frequency
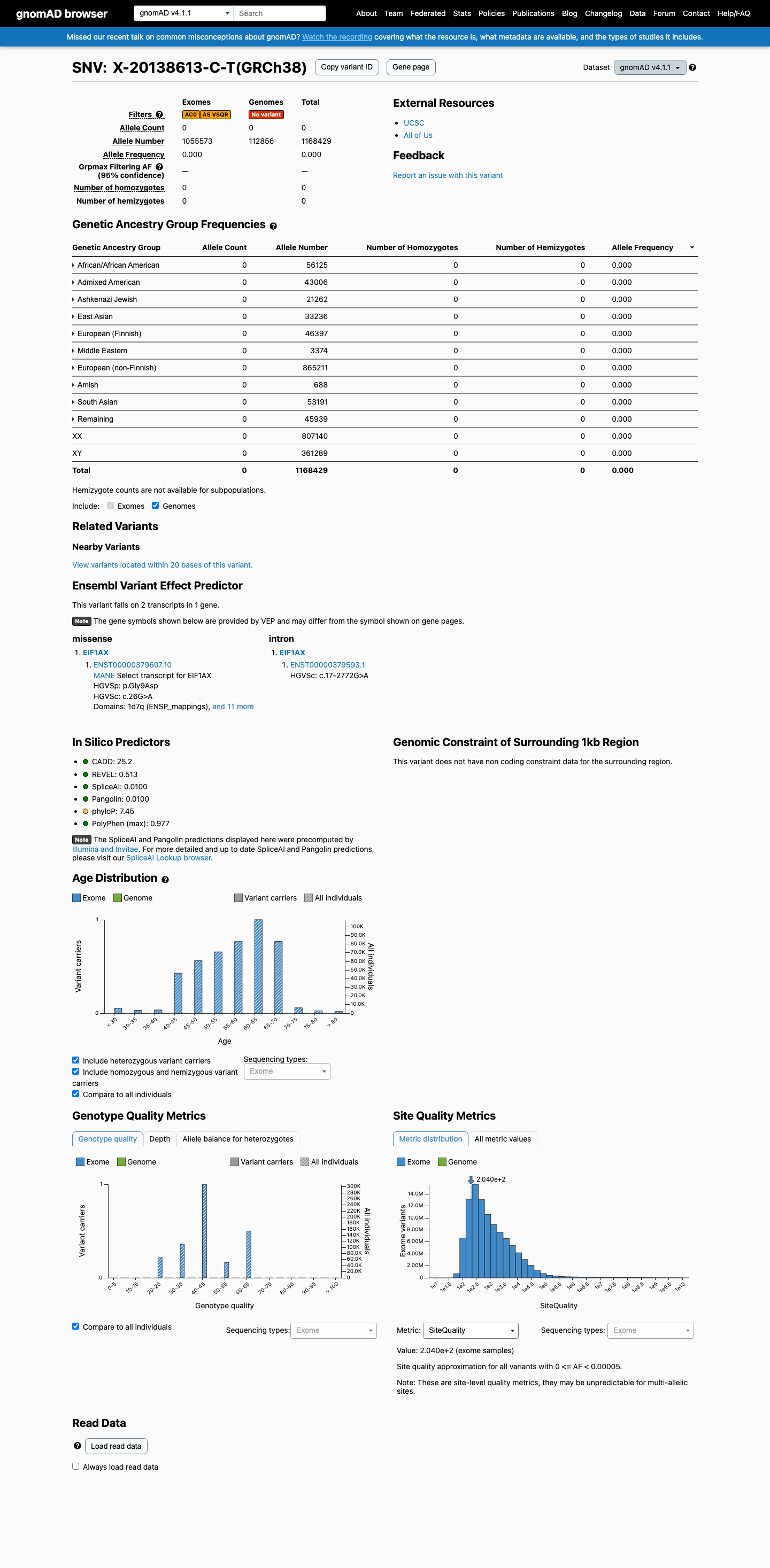
gnomAD v4.1
gnomAD v2.1
v4.1
This variant is present in gnomAD v4.1 (AF= 0; MAF= 0.00000%, 0/1168429 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 0; MAF= 0.00000%, 0/56125 alleles, homozygotes = 0).
v2.1
Absent from gnomAD v2.1.
🇨🇦 CA
Absent from gnomAD-Canada v1.0.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
Absent
· 0 / 1,168,429
0 hom
0 hom
Not observed in any ancestry group.
+ 10 not observed (Remaining individuals, Admixed American, European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish, African/African American, European (non-Finnish))
gnomAD v2.1
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
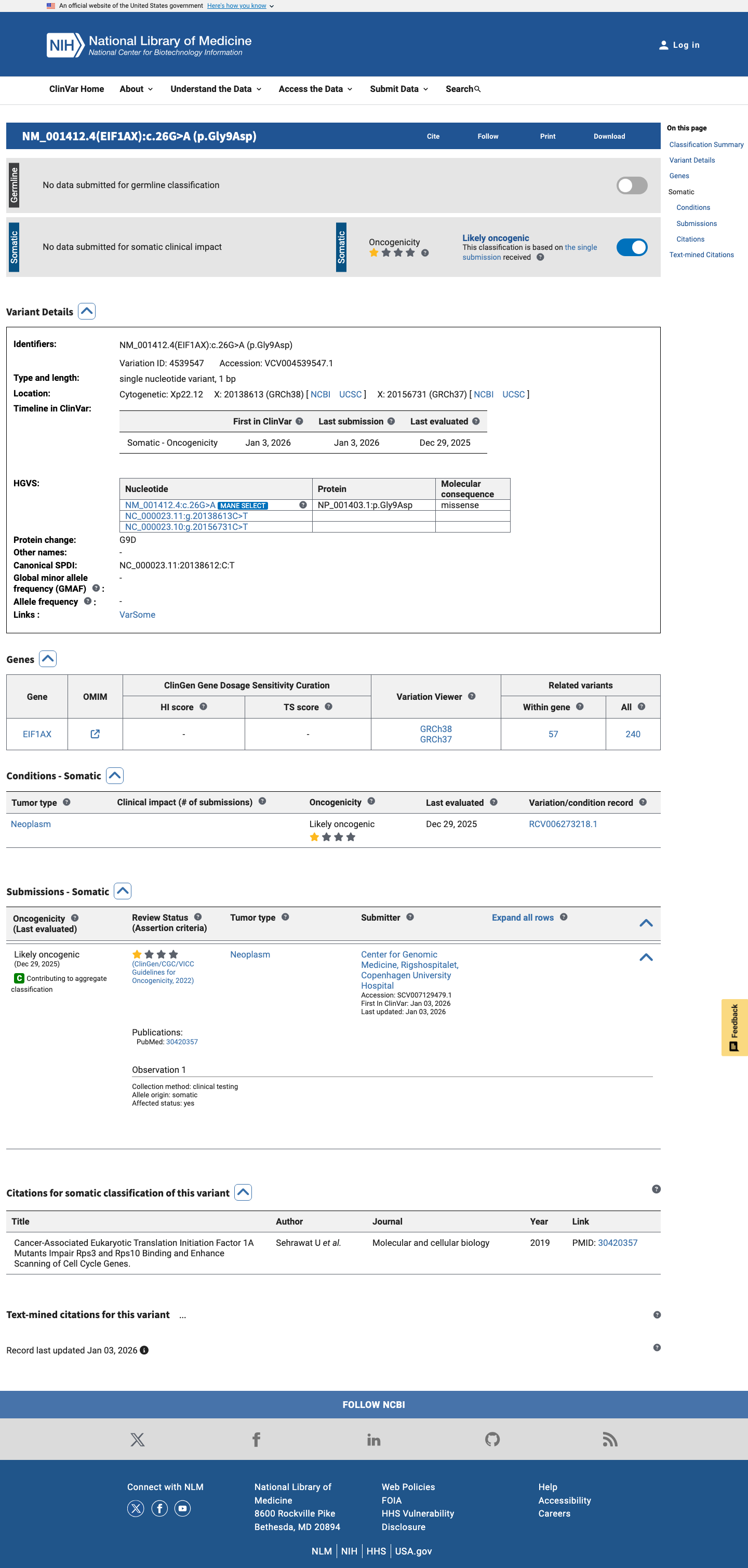
ClinVar
This variant has been reported in ClinVar but submission details could not be extracted. (ClinVarID = 4539547)
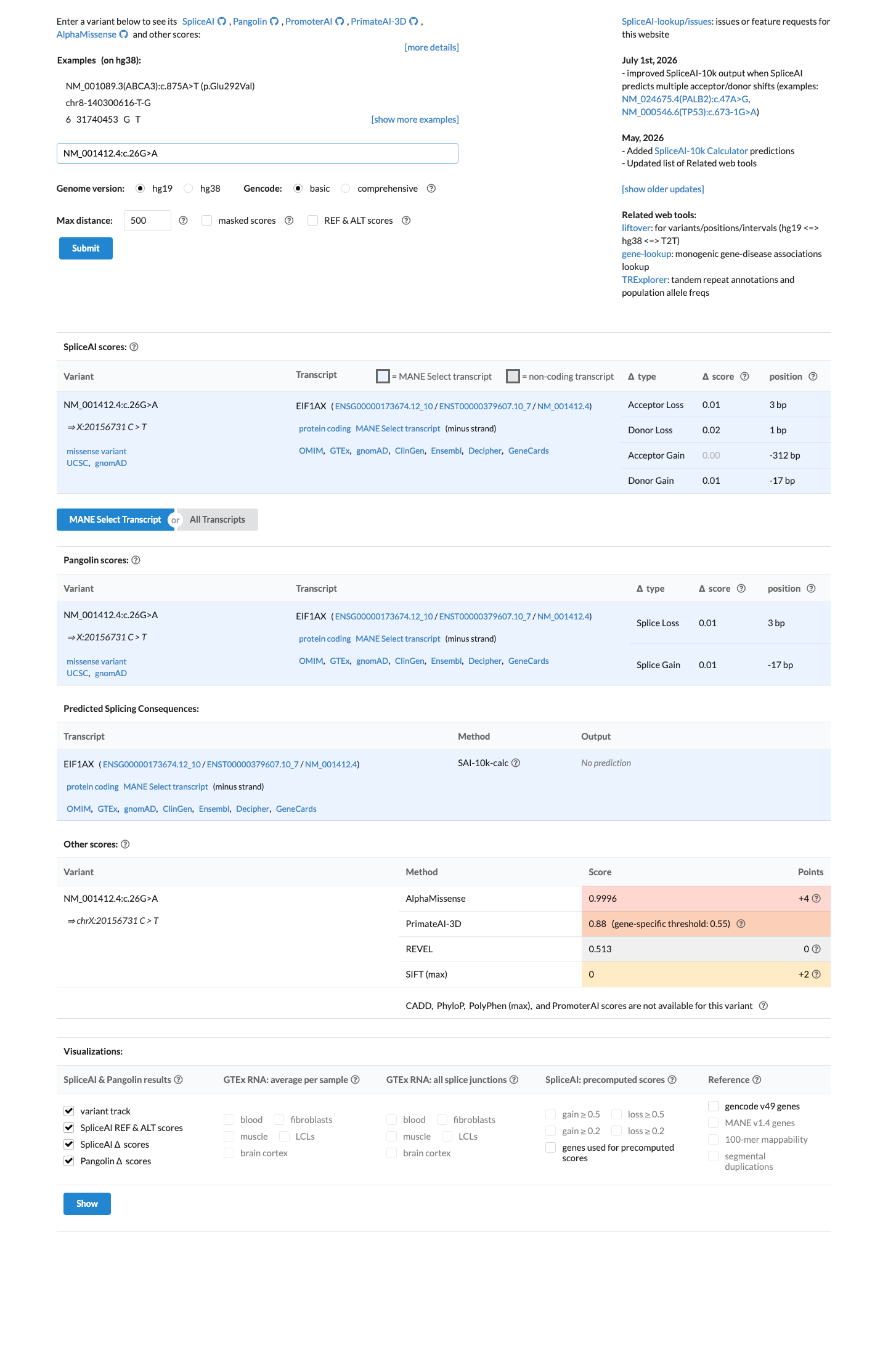
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.02). BayesDel score = -0.0342974.
Functional
Likely Oncogenic
OncoKB identified variant-specific curated literature and context relevant to functional review; biological-effect context: Likely Gain-of-function; curated oncogenicity label: Likely Oncogenic.
COSMIC
Cancer hotspots
Somatic evidence
Hotspot
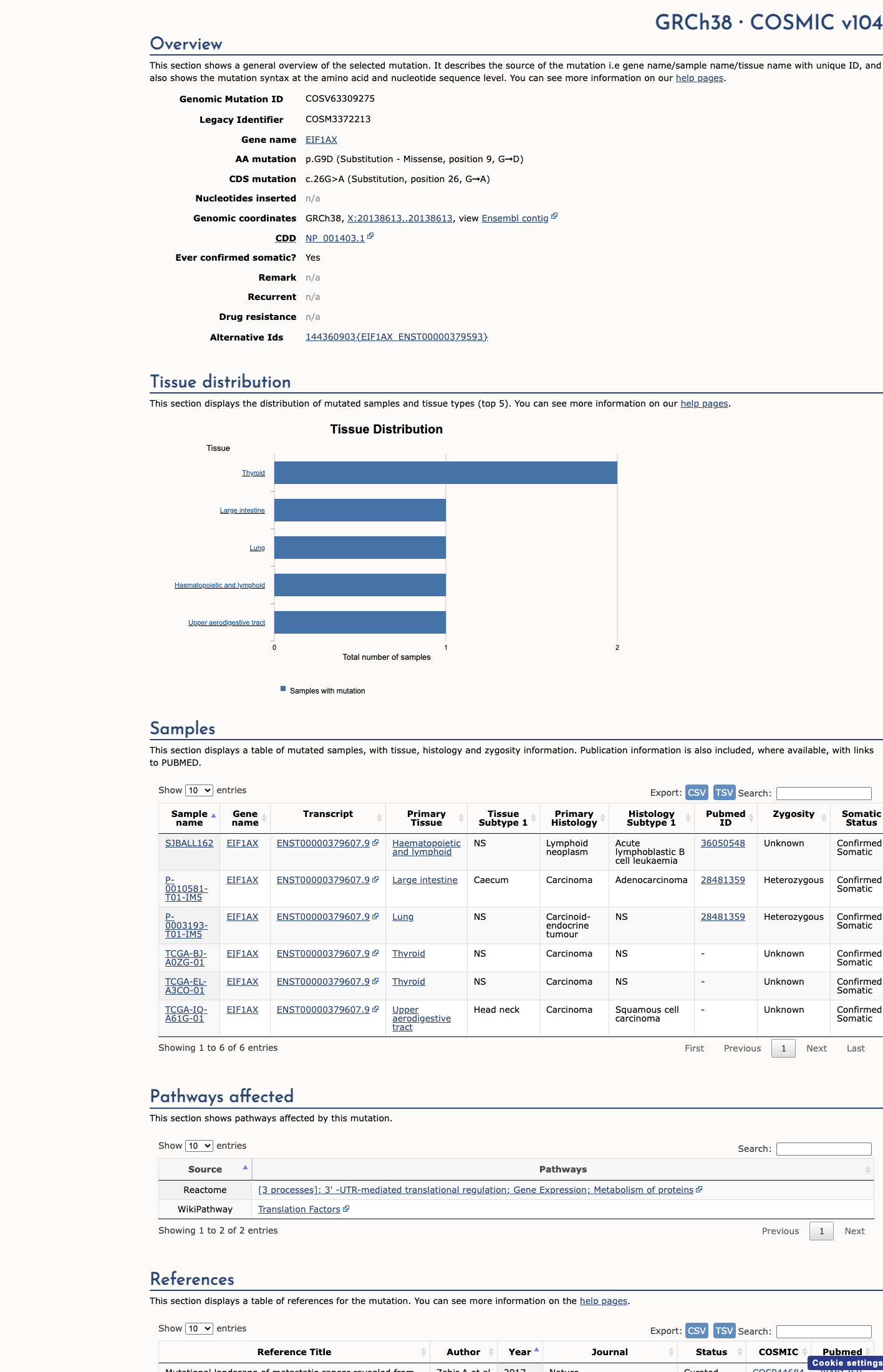
COSMIC
This variant lies in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV63309275, n = 6 times).
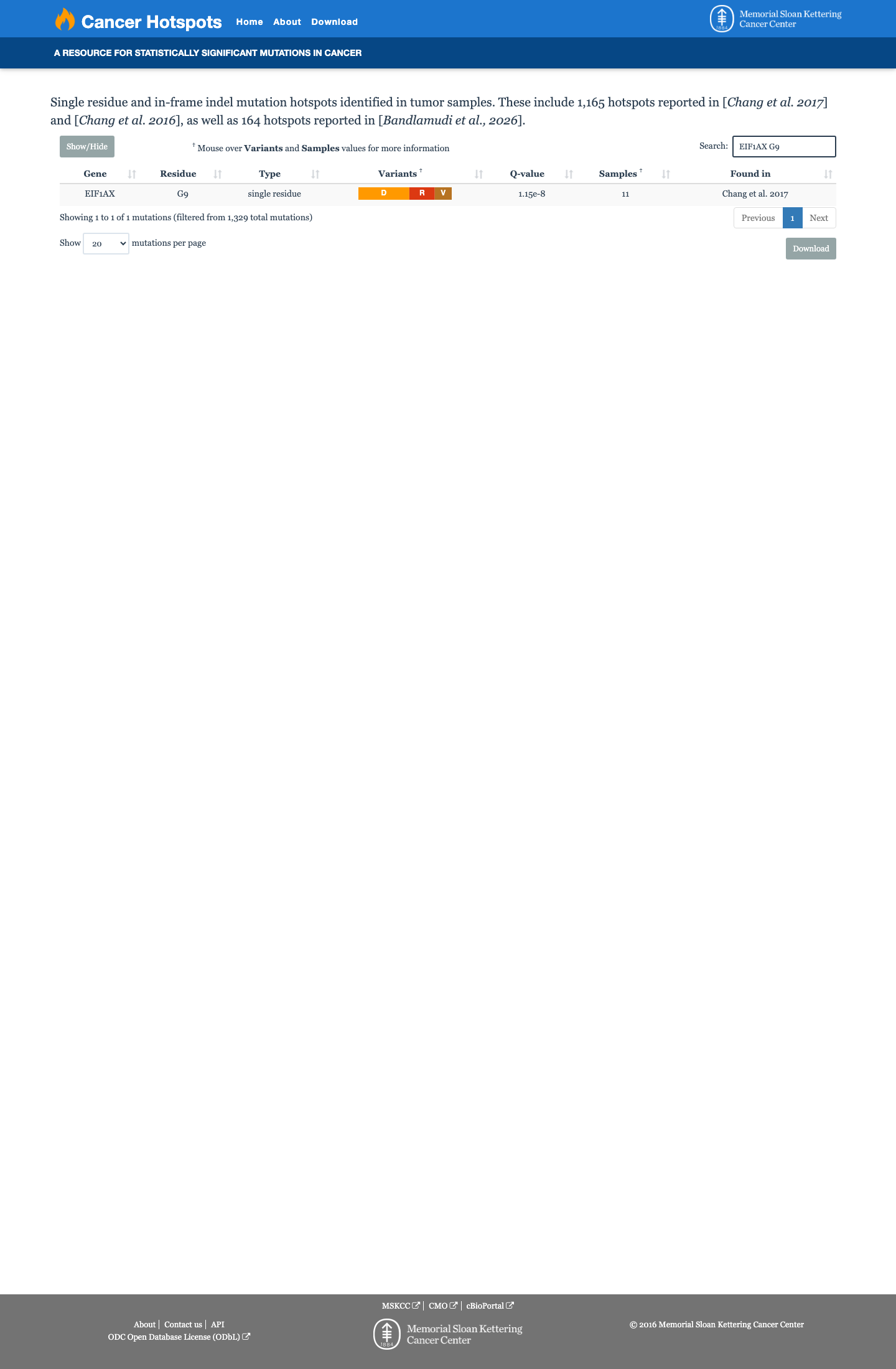
Hotspots
This variant lies in a statistically significant hotspot.
Literature · how each cited paper was used
2papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 5 further PMIDs triaged but not cited — see Sources & References.
EIF1AX and RAS Mutations Cooperate to Drive Thyroid Tumorigenesis through ATF4 and c-MYC.
Searched
G9DGly9Aspc.26G>AG9RG8RG15VNTT mutant
Found
Studied EIF1AX mutations in advanced thyroid cancers. Characterized NTT mutants G8R, G9R, and G15V, along with the A113splice CTT hotspot mutation. Demonstrated that EIF1AX NTT mutants have increased affinity for PIC components (EIF2α, EIF5) and increase global protein synthesis. EIF1AX mutations clustered within the first 15 amino acids of the NTT and cooperate with oncogenic RAS to drive thyroid tumorigenesis. G9D was not specifically studied or mentioned.
Variant
◇ Residue / gene-level — variant not named
Applied to
→PM1 supports · met
Why
G9D not directly studied. Paper provides domain-level evidence confirming the NTT as a functional hotspot for cancer mutations. Cited for PM1 (domain-level support).
NTT mutants (G8R, G9R, G15V) or EIF1AX-c'spl with an antibody to HA showed pulldown of the TC component
Location Results, paragraph 1: NTT mutants listed (G8R, G9R, G15V). Figure 3: co-IP experiments with NTT mutants. Figure 5: mTOR signaling data. · Context Co-immunoprecipitation in HEK293T cells; isogenic thyroid cancer cell lines (CAL62, TTA1, C643); Nthy-Ori 3-1 immortalized thyroid cells; mouse models (Tg-rtTA/TRE-EIF1AX-c'spl). · full text
Cancer-Associated Eukaryotic Translation Initiation Factor 1A Mutants Impair Rps3 and Rps10 Binding and Enhance Scanning of Cell Cycle Genes.
Searched
G9DGly9Aspc.26G>AG9RG9 DNTT mutant
Found
EIF1AX G9D single mutant was generated by site-directed mutagenesis alongside six other NTT mutants. Functional characterization was performed on the G9D/R13H double mutant, which showed reduced binding to ribosomal protein Rps10 in in vitro pull-down assays (not significant for Rps3). The G9R single mutant (same position) showed enhanced translation of long 5'UTR mRNAs in luciferase reporter assays. All seven NTT cancer-associated mutants displayed enhanced scanning compared to wild-type, consistent with a gain-of-function mechanism impairing scanning arrest.
Variant
✓ Names this variant — characterised directly
Applied to
→PM1 supports · met
→PS3 supports · met
Why
G9D single mutant was generated but standalone functional data not shown. G9D/R13H double mutant data and same-position G9R data support gain-of-function effect; cited for PS3 at supporting strength and PM1 domain-level evidence.
mutants (G6D, G9D, G9R, G8RG9R, K10E, R13H and G15D) were generated by site directed mutagenesis
Location Methods (Plasmids section): G9D listed as one of seven NTT mutants generated. Results (Figure 8): G9D/R13H double mutant tested in Rps3/Rps10 binding assays. Figure 7: G8R/G9R and other NTT mutants tested in luciferase reporter and polysome profiling assays. · Context In vitro binding assays using bacterially expressed His-tagged Rps3/Rps10 with untagged eIF1A mutants; luciferase reporter assays in MEF and HEK293T cells; polysome profiling in HEK293T cells. · full text
Sources & reference links
Triaged references · 5 PMIDs not cited in assessment
26878173 ↗
Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers.
ONCOKB
35101336 ↗
Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC).
CLINVAR
22918138 ↗
Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology.
CLINVAR
34131312 ↗
Chromosomal microarray analysis, including constitutional and neoplastic disease applications, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG).
CLINVAR
23619274 ↗
American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders.
CLINVAR