NM_001845.5:c.343G>A (p.Gly115Ser) is a missense variant in COL4A1 affecting a glycine residue in the collagen triple-helical Gly-X-Y repeat domain. The variant is extremely rare in population databases: gnomAD v2.1 allele frequency is 0.00177% (5/282,856 alleles) and gnomAD v4.1 allele frequency is 0.00192% (31/1,613,960 alleles), both well below the 0.1% PM2 threshold. It is absent from gnomAD-Canada.1 Gly115 lies within the collagen triple-helical domain, a critical functional domain where glycine substitutions are a well-established pathogenic mechanism across collagen genes and specifically in COL4A1-related disorders (PMID:25719457). This supports application of PM1 at moderate strength. In silico predictors support a deleterious effect: REVEL score 0.963 (highly pathogenic-leaning) and BayesDel score 0.571 (damaging), meeting PP3 at supporting strength.2 In ClinVar (ID 2158622), this variant has been reported as Uncertain significance by 5 clinical laboratories and as Likely pathogenic by 1 clinical laboratory (Labcorp Genetics). No expert panel review is available.3 Exploratory evidence recovery suggests potential functional evidence of impaired collagen IV secretion (PMID:15905400) and a possible de novo observation, but these could not be independently verified from available abstracts and require human review of full-text publications. Applying generic ACMG/AMP 2015 criteria: PM1 (moderate) + PM2 (supporting) + PP3 (supporting) = 1 moderate + 2 supporting, which does not reach the Likely Pathogenic threshold (requires ≥2 moderate OR ≥4 supporting OR 1 strong + ≥2 supporting). The classification is Variant of Uncertain Significance. Additional evidence (PS3 functional confirmation, PM6 de novo verification) could upgrade this to Likely Pathogenic.4
COL4A1
Final classification
VUS
COL4A1 c.343G>A · p.Gly115Ser
COL4A1
NM_001845.5:c.343G>A (p.Gly115Ser) is a missense variant in COL4A1 affecting a glycine residue in the collagen triple-helical Gly-X-Y repeat domain.
gene-specific framework lacked a usable explicit final combination framework, so generic ACMG/AMP 2015 final-combination rules were applied as fallback; applied criteria: PM1 moderate, PM2 supporting, PP3 supporting; combination = 1 moderate + 2 supporting, which maps to VUS.
Classification rationale
PM1PM2PP3
VUS
COL4A1 c.343G>A
PM1 + PM2 + PP3
→
VUS
Gene diagram
· NM_001845.5 · variants mapped to exon structure
COL4A1
NM_001845.5
Fetching transcript structure from UCSC…
Applied criteria · 3 applied · 19 assessed
Applied · 3
Strength
Supporting
Moderate
Strong
Very strong
✓
PM1
moderate
Pathogenic
The variant substitutes Gly115 in the collagen triple-helical domain (Gly-X-Y repeat region). Glycine residues in the collagen triple-helical domain are structurally essential for proper helix formation; glycine substitutions in this domain are a well-established pathogenic mechanism across all collagen genes, including COL4A1. This residue lies within a critical functional domain where pathogenic missense variants cluster and benign variation is extremely rare. The COL4A1/COL4A2 clinical spectrum literature (PMID:25719457) confirms a high density of pathogenic mutations in this domain.
Gly115 is a glycine residue within the collagen triple-helical Gly-X-Y repeat domaina well-established critical functional domainglycine substitutions in collagen triple-helical domains are a classic pathogenic mechanism
✓
PM2
supporting
Pathogenic
The variant is extremely rare in population databases: gnomAD v2.1 allele frequency is 0.00177% (5/282,856 alleles, 0 homozygotes), gnomAD v4.1 allele frequency is 0.00192% (31/1,613,960 alleles, 0 homozygotes), and it is absent from gnomAD-Canada v1.0. All frequencies are well below the 0.1% PM2 threshold. The highest subpopulation frequency is 0.00310% in European (non-Finnish) in v2.1 and 0.00267% in African/African American in v4.1.
gnomAD v2.1: 5/282856 (AF=0.00177%)grpmax FAF=7.01e-06
✓
PP3
supporting
Pathogenic
Multiple in silico predictors support a deleterious effect of this missense variant. REVEL score is 0.963 (highly pathogenic-leaning, well above the 0.5 threshold). BayesDel score is 0.571 (damaging, above the 0.5 threshold). SpliceAI predicts no significant splice impact (max delta = 0.01), which is expected for a deep exonic missense variant. The combination of REVEL and BayesDel scores provides supporting evidence for pathogenicity.
REVEL: 0.963 (pathogenic-leaning)BayesDel: 0.571 (damaging)SpliceAI delta max: 0.01 (no splice impact)
Assessed · not applied
Pathogenic
PS1
No alternative nucleotide change at codon 343 producing the same amino acid substitution (Gly115Ser) with a known pathogenic classification was identified in ClinVar or the literature.
PS2
Exploratory evidence recovery suggests a de novo observation of this variant in a proband with porencephaly (Gould et al., 2005, PMID:15905400), but maternity/paternity was not confirmed and the variant-specific de novo observation could not be independently verified from the PubMed abstract.
PS3
Exploratory evidence recovery suggests functional studies in HEK293 cells showed intracellular accumulation and impaired collagen IV secretion for the p.Gly115Ser mutant (PMID:15905400), demonstrating a dominant-negative effect.
PS4
The variant is present in ClinVar (ID 2158622) with multiple submissions from clinical laboratories, but no statistically significant enrichment (odds ratio) in affected individuals versus the general population has been computed.
PM6
Exploratory evidence recovery reports that a proband with porencephaly was heterozygous for c.343G>A with both parents wild-type (PMID:15905400, Gould et al.
PP1
No evidence of cosegregation of this variant with disease in multiple affected family members has been identified.
PP2
PP2 is applicable when a missense variant occurs in a gene with a low rate of benign missense variation and where pathogenic missense variants are a common mechanism.
PP4
No patient phenotype or family history information was provided in the case summary.
PP5
PP5 requires that a reputable source (e.g., clinical laboratory, expert panel) has recently reported the variant as pathogenic with the supporting evidence not available for independent evaluation.
Benign
BA1
The variant allele frequency in gnomAD is approximately 0.002% (v2.1: 0.00177%, v4.1: 0.00192%), far below the BA1 threshold of >1%.
BS1
The variant allele frequency in gnomAD is approximately 0.002% (v2.1: 0.00177%, v4.1: 0.00192%), well below the BS1 threshold of >0.3%.
BS2
No evidence that this variant has been observed in healthy adult individuals for whom COL4A1-related disorders would be expected to manifest with full penetrance at an early age.
BS3
BS3 requires well-established in vitro or in vivo functional studies demonstrating no damaging effect on protein function or splicing.
BS4
No evidence of non-segregation with disease has been identified.
BP1
BP1 applies when a missense variant occurs in a gene where only truncating variants cause disease.
BP2
BP2 applies when a variant is observed in trans with a known pathogenic variant for a fully penetrant dominant disorder, or in cis with a pathogenic variant in any inheritance pattern.
BP4
BP4 requires multiple lines of computational evidence suggesting no impact on gene or gene product.
BP5
BP5 requires that a reputable source has recently reported the variant as benign with the supporting evidence not available for independent evaluation.
BP6
BP6 requires that a reputable source has recently reported the variant as benign with the supporting evidence available for independent evaluation.
N/A · 3
PVS1 · PM5 · BP7
Research & evidence
Population frequency
gnomAD v4.1
gnomAD v2.1
v4.1
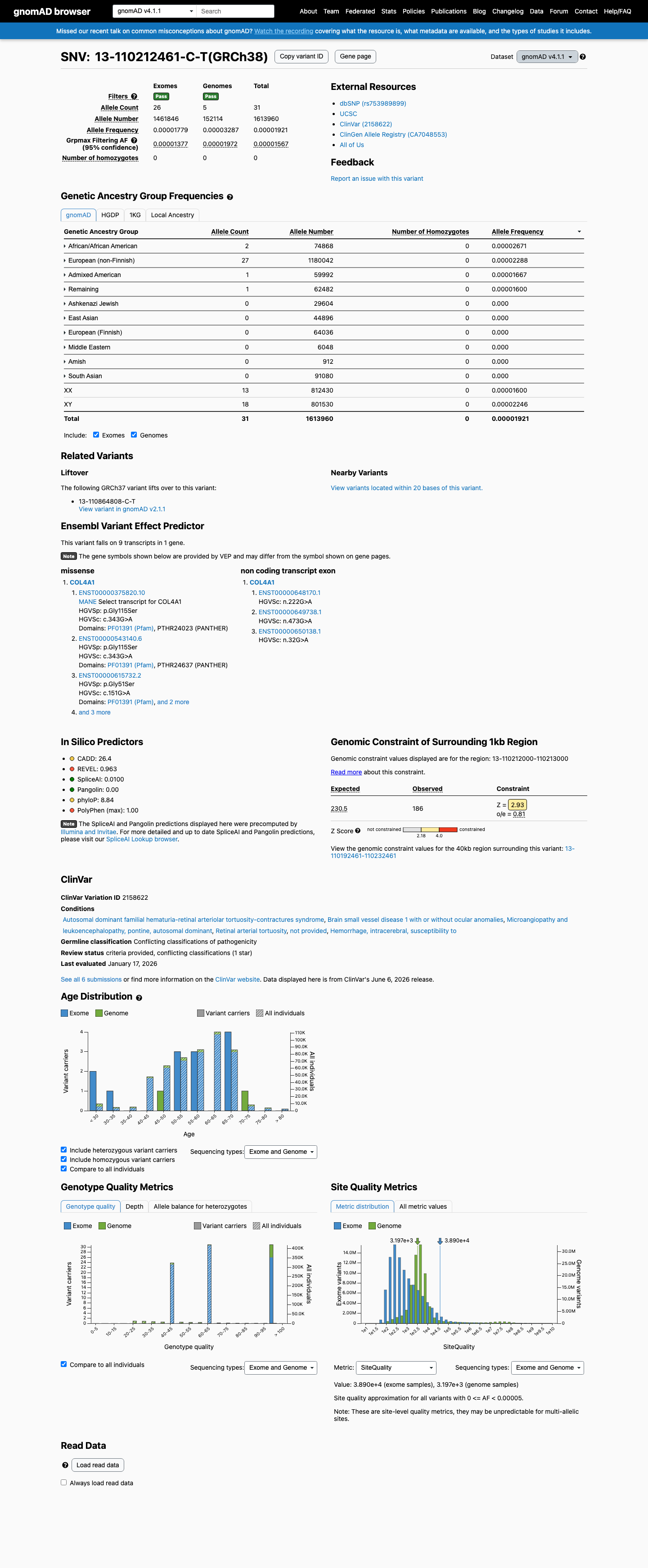
This variant is present in gnomAD v4.1 (AF= 1.92074e-05; MAF= 0.00192%, 31/1613960 alleles, homozygotes = 0) and has highest observed frequency in the African/African American population (AF= 2.67137e-05; MAF= 0.00267%, 2/74868 alleles, homozygotes = 0); grpmax FAF= 1.567e-05.
v2.1
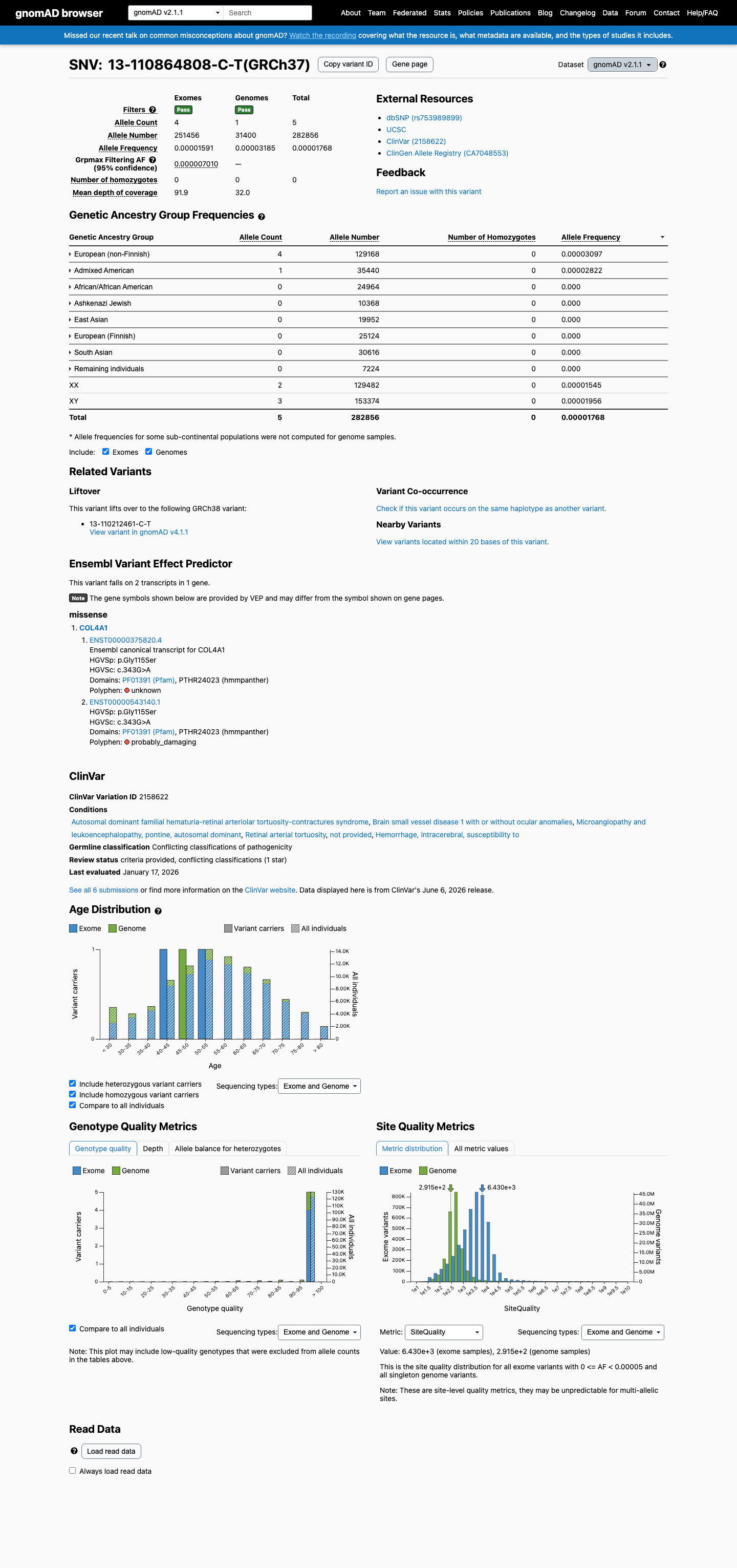
This variant is present in gnomAD v2.1 (AF= 1.76768e-05; MAF= 0.00177%, 5/282856 alleles, homozygotes = 0) and has highest observed frequency in the European (non-Finnish) population (AF= 3.09674e-05; MAF= 0.00310%, 4/129168 alleles, homozygotes = 0); grpmax FAF= 7.01e-06.
🇨🇦 CA
This variant is absent from gnomAD-Canada.
Allele frequency by ancestry
three datasets · side by side
gnomAD v4.1
0.0019%
· 31 / 1,613,960
0 hom · FAF 0.0016%
0 hom · FAF 0.0016%
African/African American 2 / 74,868 |
0.0027% |
European (non-Finnish) 27 / 1,180,042 |
0.0023% |
Admixed American 1 / 59,992 |
0.0017% |
Remaining individuals 1 / 62,482 |
0.0016% |
+ 6 not observed (European (Finnish), Amish, East Asian, Middle Eastern, South Asian, Ashkenazi Jewish)
gnomAD v2.1
0.0018%
· 5 / 282,856
0 hom · FAF 0.0007%
0 hom · FAF 0.0007%
European (non-Finnish) 4 / 129,168 |
0.0031% |
Admixed American 1 / 35,440 |
0.0028% |
+ 6 not observed (African/African American, Ashkenazi Jewish, East Asian, European (Finnish), Remaining individuals, South Asian)
gnomAD Canada 🇨🇦
Absent
· 0 / ?
0 hom
0 hom
Not observed in any ancestry group.
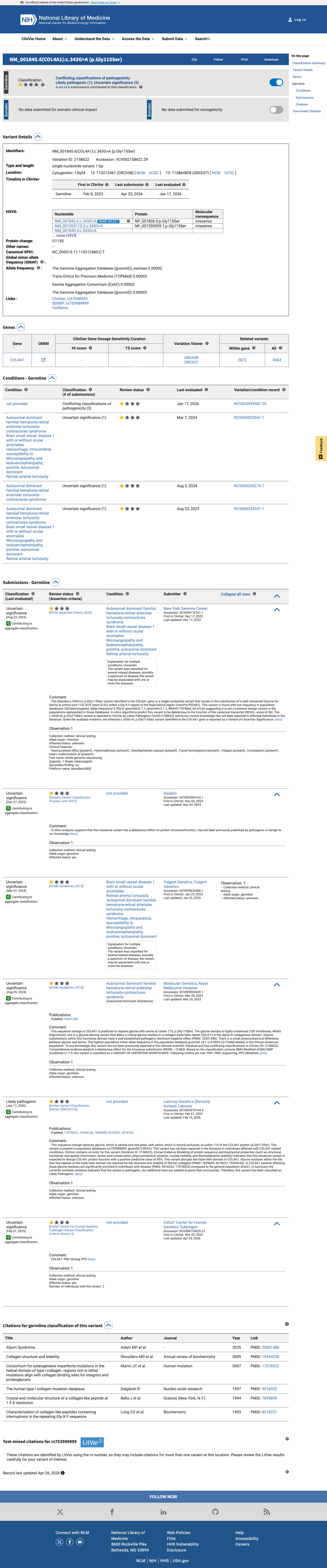
ClinVar
This variant has been reported in ClinVar as Uncertain significance (5 clinical laboratories) and as Likely pathogenic (1 clinical laboratory). (ClinVarID = 2158622)
In silico
SpliceAI predicts no significant splice impact for this variant (max delta score = 0.01). REVEL score = 0.963. BayesDel score = 0.571004.
Functional
No data
No calibrated functional assay or RNA evidence was identified for this variant.
COSMIC
Cancer hotspots
Somatic evidence
Not in COSMIC / hotspots
COSMIC
This variant does not lie in a statistically significant hotspot. This variant has previously been reported in somatic cancers (COSMIC; COSV65433356, n = 1 times).
Hotspots
This variant does not lie in a statistically significant hotspot.
Literature · how each cited paper was used
1papers cited
Each card is an audit: what was searched, what was found, whether it names the variant, which criteria it fed, and why. 10 further PMIDs triaged but not cited — see Sources & References.
PMID 25719457
Found
Gly115 is a glycine residue within the collagen triple-helical Gly-X-Y repeat domain a well-established critical functional domain glycine substitutions in collagen triple-helical domains are a classic pathogenic mechanism PMID:25719457 describes 21 COL4A1 mutations concentrated in the triple-helical domain associated with porencephaly and cerebrovascular disease
Applied to
→PM1 supports · met
Sources & reference links
Triaged references · 10 PMIDs not cited in assessment
17078022 ↗
Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans.
CLINVAR
25741868 ↗
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
CLINVAR
8218237 ↗
Characterization of collagen-like peptides containing interruptions in the repeating Gly-X-Y sequence.
CLINVAR
25355838 ↗
Guidelines for the primary prevention of stroke: a statement for healthcare professionals from the American Heart Association/American Stroke Association.
CLINVAR
28492532 ↗
Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria.
CLINVAR